





Abstract
It has long been assumed that β-amyloid (Aβ) had to assemble into fibrillar amyloid plaques to exert its neurotoxic effects in Alzheimer disease. An alternative hypothesis is that soluble oligomers ofAβ play a much larger role in neuronal damage than the insoluble component. We have tested these competing hypotheses in vivo by studying the clinicopathologic correlates of oligomeric Aβ species and classic fibrillar amyloid plaques in the brains of double-transgenic APPsw-tauvlw mice up to 17 months of age. Biochemical and immunohistochemical measures of brain oligomeric Aβ exponentially increased with age. Oligomeric Aβ load correlated with morphological markers of fibrillar Aβ deposition. In contrast to total amyloid plaque burden, the amount of oligomeric Aβ deposits labeled by the conformational epitope-specific antibody Nab61 closely correlated with neuronal loss and numbers of astrocytes in the entorhinal cortex and the CA1 hippocampal subfield. However, like other morphological Aβmeasurements, brain oligomeric Aβ burden did not correlate well with memory deficits in these mice. The number of glial fibrillary acidic protein-positive astrocytes in entorhinal cortex and CA1 most tightly correlated with memory impairment and neuronal cell loss. Based on these findings, we hypothesize that the astrocyte response, which is likely triggered by brain oligomeric Aβ accumulation, adversely affects cognition and might also contribute to neuronal cell death in this model.
Introduction
In recent years, the idea that brain accumulation of β-amyloid (Aβ) is the primary influence that triggers the cascade of pathogenic events in Alzheimer disease (AD) has become the leading pathogenic hypothesis (1). According to this theory, neurofibrillary tangle (NFT) formation, inflammatory changes, and the loss of neurons and synapses in vulnerable regions that invariably accompany Aβ deposition in the AD brain are downstream events resulting from accumulation of Aβ. Yet, this hypothesis remains controversial mainly because the specific neurotoxic species of Aβ and its effects on neuronal function in vivo have not been defined. It has been long assumed that Aβ had to be assembled into extracellular amyloid fibrils (amyloid plaques) to exert its cytotoxic effects; however, multiple clinicopathologic studies have been unable to confirm a relationship between amyloid plaque load and dementia severity or loss of neurons and synapses (2-4). Moreover, numerous amyloid plaques that match the brain regional pattern of distribution seen in demented AD patients may also be present in many nondemented individuals (5-7). These data raise a central paradox in the field of AD neuropathology: if amyloid is the key molecule, why does it not correlate better with clinical manifestations or neuronal and synaptic loss?
A second hypothesis is that the plaques themselves are not directly toxic but, instead, they lead to another toxic species, soluble oligomeric forms of Aβ, which play a larger role than the insoluble components in synaptic and neuronal damage in AD (8). In support of this alternate hypothesis are data showing that soluble forms of Aβ correlate more closely with dementia severity than fibrillar Aβ (9-11). It has recently been shown that stable Aβ oligomers decrease cell surface expression of N-methyl-D-aspartic acid-type receptors, inhibit induction of long-term potentiation, facilitate long-term depression, alter dendritic spine density, and affect hippocampal synaptic plasticity (12-19). Together, these studies suggest that oligomeric forms of Aβ are potentially responsible for loss of connectivity and disrupted memory-related synaptic plasticity in AD. However, several fundamental questions remain unanswered. Specifically, does oligomeric Aβ correlate with other measures of Aβ deposition (classical fibrillar amyloid plaques), and how does oligomeric Aβ relate to the other neurodegenerative hallmarks of AD (e.g. tau pathology, neuronal cell loss, and gliosis) and with clinical manifestations?
Transgenic (Tg) mice have provided important insights into the chronology of events leading to the fibrillar amyloid plaque deposition, gliosis, NFTs, and neuronal cell death that characterize AD. For example, double-Tg mice that overexpress human mutant amyloid precursor protein (APP) and tau (Tg line APPsw-tauvlw) mimic several features of the AD phenotype to a remarkable extent, including amyloid deposition, abnormal tau phosphorylation/NFT-like formation, glial cell proliferation, and significant neuronal loss in selectively vulnerable brain regions, for example, the entorhinal cortex (EC) and CA1 subfield of the hippocampus (20, 21). All the above phenotypic traits in these mice develop following an age-dependent fashion and are accompanied by progressive hippocampus-dependent memory impairment. Interestingly, neuronal cell death in these mice predates overt amyloid plaque deposition, arguing against a direct major toxic effect of extracellular fibrillar amyloid on neuronal survival. As in AD brains, the amount of neuronal loss in these mice does not correlate well with total immunostained amyloid plaque burden (21). Thus, these mice give us an excellent opportunity to track the natural history of oligomeric Aβ accumulation in their brains and to study the relationships of these Aβ species to AD-related neuropathological changes and cognition.
We measured oligomeric Aβ species and classic fibrillar amyloid plaques in the brain of APPsw-tauvlw Tg mice through 17 months of age and evaluated their relation with memory performance, astroglial cell proliferation, and number of neurons in the EC, CA1, and cingulate cortex. Our results showed that accumulation of soluble oligomeric forms of Aβ in the brain of these animals was an early event that preceded fibrillar amyloid deposition, astrogliosis and neuronal loss, and significantly increased with age. NAB61, an antibody that recognizes a conformational epitope present in dimeric, small oligomeric and higher order Aβ structures (22), recognized a subset of brain Aβ deposits, preferentially mature senile plaques, and their amount correlated with other morphological markers of fibrillar Aβ deposition. Oligomeric Aβ deposits first appeared in the EC and the CA1, faithfully matching the selective regional pattern of neuronal vulnerability observed in these mice and, as opposed to total Aβ plaque burden, were tightly correlated with the amount of neuronal loss and reactive astrocytes. However, like other morphological markers of Aβ deposition, brain oligomeric Aβ load did not correlate well with memory performance. Interestingly, the number of glial fibrillary acidic protein (GFAP)-positive astrocytes was the parameter most closely correlated with memory impairment and neuronal loss. Based on our findings, which argue against the idea that inflammation is merely a bystander in AD, we hypothesize that the inflammatory response of astroglial cells, likely triggered by brain oligomeric Aβ accumulation, may be a key event in memory disruption and neuronal cell death in this Tg mouse model, and that this might also be relevant in AD.
Materials and Methods
Animals
Tg APPsw-tauvlw mice overexpressing human mutant APP (Swedish mutation K670N-M671L) and a triple human tau mutation associated with frontotemporal dementia and parkinsonism linked to chromosome 17 (G272V, P301L, and R406W) on a mixed hybrid genetic background (C57Bl6j/SJL/CBA) (20, 21) were used. In brief, at 9 months of age, these mice begin to exhibit scarce amyloid deposits and infrequent tau filament formation in limbic and cortical areas, as well as incipient neuronal loss in the EC, without significant memory impairment. Severe abnormalities, including abundant amyloid plaque deposition, increased levels of tau phosphorylation and aggregation (but no mature NFT formation), glial cell proliferation, and pronounced neuronal loss in the EC and the CA1 region of the hippocampus can be seen in mice at 16 months, along with overt hippocampal-dependent memory deficits. Mature argyrophilic NFT formation is a late event that can be demonstrated at approximately 25 months of age in the EC and CA1, matching the regional pattern of neuronal vulnerability observed at earlier ages.
Groups of Tg APPsw-tauvlw mice (n = 26) and age and gender-matched wild-type (wt) littermate controls (n = 26) were studied at different ages ranging from 5 to 17 months. All experiments were conducted in accordance to our institutional Animal Care and Use Committee guidelines and conformed to the European Union Directive 86/609/EEC.
Spatial Reference Learning and Memory Testing
Mice underwent spatial reference learning and memory testing in the Morris water maze, according to previously described protocols (23). In brief, the maze was a circular pool (diameter 1.5 m) filled with water at 20°C. The mice underwent visible-platform training for 3 consecutive days (8 trials/day), using a platform raised above the water. This was followed by hidden-platform training, during which the mice were trained to locate a platform submerged 1 cm beneath the surface for 9 consecutive days (4 trials/day). Each trial was terminated when the mouse reached the platform or after 60 seconds, whichever came first. The average escape latency throughout the 9 days of training was calculated for each mouse. At 24 hours after the 12th, 24th, and 36th trials, mice were subjected to a probe trial in which they swam for up to 60 seconds in the pool with no platform. The average percentage of time spent in the correct quadrant of the pool throughout the 3 probe trials was calculated for each mouse. Trials were recorded using an HVS water maze program for analysis of swimming speed, escape latencies, and percent of time spent in each quadrant of the pool during probe trials (analysis program Video-Tracking SMART, Panlab, Barcelona, Spain). All mice were tested in a coded manner.
Contextual Fear Conditioning
For contextual fear conditioning, the mice were placed within the conditioning chamber and allowed to explore it for 3 minutes before the onset of the unconditioned stimulus, foot shock (1 second/1 mA). After the foot shock, they were left in the chamber for 2 minutes (immediate freezing) and returned to their home cages. Conditioning was tested 24 hours after training for 4 minutes in the same conditioning chamber. Freezing response (a behavioral index of conditioned fear response) was scored using an automated video tracking fear conditioning system (Med Associates, St. Albans, VT).
All animals underwent screening for the presence of a naturally occurring mutation in several strains of mice, including the SJL strain that causes retinal degeneration because of the inactivation of the PDE6 gene, according to previously published protocols (24). Adult mice homozygous for the mutation are known to be blind and were excluded from behavioral tests.
Neuropathology
Tissue Preparation
Mice were killed under isoflurane administration, and brains were immediately removed. One hemisphere was snap frozen in dry ice for Dot Blot and enzyme-linked immunosorbent assay (ELISA). The other hemisphere was fixed for 24 hours in 4% paraformaldehyde in phosphate buffered saline (PBS), pH 7.4, and coronally sectioned at 30 μm on a freezing sledge microtome for detailed quantitative histological analyses.
Immunohistochemistry
Thirty-micrometer-thick coronal sections were permeabilized with 0.5% Triton-X100 in PBS, blocked with normal goat serum, and sequentially probed with primary antibody (4G8 mouse anti-Aβ 1:500 [Chemicon, Temecula, CA]; mouse Nab61 1:100, a kind gift of Dr. Virginia Lee [University of Pennsylvania]; CP13 1:50 mouse anti-Ser-202 phospho-tau and PHF-1 1:50 mouse anti-Ser-396/404 phospho-tau, kind gifts of Dr. Peter Davies [Yeshiva University]; rabbit anti-GFAP 1:500 [Chemicon]; NeuN 1:200 [Chemicon]; anti-NF-κB p105/p50 1:100 [Cell Signaling, San Diego, CA]) and the appropriate secondary antibody (anti-mouse and anti-rabbit IgG 1:200 [Southern Biotechnology, Birmingham, AL], Vector ABC Kit [Vector Laboratories, Burlingame, CA]). Sections were also processed by thioflavin-S (ThioS) staining for dense-core plaque counts.
Amyloid Burden Quantification
Amyloid deposition was quantified using Aβ immunostaining (monoclonal anti-Aβ 4G8 and horseradish peroxidase-anti-mouse) and the Bioquant Nova Prime V6 90.10 system, according to previously published protocols (3, 25). In brief, video images of each region of interest in 30-mm-thick sections were captured, and a threshold of optical density that discriminated staining from background was obtained. Manual editing eliminated artifacts. The total “amyloid burden” was defined as the total percentage of cortex covered by immunostained amyloid deposits over 3 sections and was calculated for EC, CA1, and cingulate cortex. Identical measurements were conducted for the subpopulation of dense core plaques on ThioS-stained sections. Brain oligomeric Aβ deposition was quantified using NAB61 antibody, following identical protocols.
Stereological Astrocyte and Neuronal Counts
Astrocyte and neuronal counts were performed in EC, CA1, and cingulate cortex using the optical disector technique (26) in 30-μm-thick GFAP (astrocytes)- and NeuN (neurons)-immunostained coronal sections at equally spaced intervals (450 μm), excluding cells in the superficial plane of section. The entire volume of each region of interest was estimated according to the principle of Cavalieri, using the C.A.S.T. Grid System (Olympus).
The margins of the lateral EC were defined as follows: caudal, Bregma-3.88; rostral, Bregma-3.16; lateral, rhinal fissure; and medial, amygdalopiriform transition area. The CA1 hippocampal subfield was sampled from its caudal extent anteriorly (Bregma-3.40 to Bregma-1.46). The cingulate cortex was sampled in a caudal-to-rostral orientation: caudal, section containing the most caudal extent of the dentate gyrus (Bregma-3.64); rostral, extending anteriorly 0.94 mm (Bregma-2.70); medially, presubiculum; and laterally, visual cortex. The total number of neurons and astrocytes were estimated using 60 optical disectors in each case.
The average coefficient of error from the sampling technique was less than 0.05, suggesting that a minimal amount of variance observed in the counts is caused by variance from the technique.
Oligomeric Aβ Dot Blot and ELISA Preparation of Protein Extracts
Sixty-milligram frozen dissected mouse cortices were homogenized in 10 volumes of Tris buffered saline (TBS) with protease inhibitor cocktail (Roche, Basel, Switzerland) using a mechanical homogenizer with a microtube disposable pestle and cordless motor (Kontes, Fisher Scientific). Lysates were centrifuged at 260,000 × g for 20 minutes at 4°C, and the supernatant was used as TBS-soluble fraction (27). APPsw-tauvlw and age-matched wt mice (n = 12 each) were used for these analyses. A group of 9 sex- and age-matched mice from their parental APP line Tg2576 were used for comparison in the ELISA assessments of oligomeric Aβ.
Oligomeric Aβ Dot Blot Analysis
The TBS-soluble fraction protein (0.12 mg/mL) was loaded on nitrocellulose membranes (Schleicher & Schuell, Potran, pore size 0.2 mm). After blocking with 5% nonfat milk solution for 1 hour, membranes were probed overnight with the appropriate primary antibodies (mouse NAb61 1:10000 anti-oligomeric Aβ, mouse CP13 1:10000 mouse anti-Ser202 phospho-tau). Membranes were incubated with secondary antibody, anti-mouse IgG horseradish peroxidase conjugated for 1 hour, 1:200 (Bio-Rad Laboratories, Hercules, CA), and developed by adding detection solution (ECL Western blotting detection reagent from Amersham, UK) in a 1:1 ratio for solution 1:solution 2. The blots were quantified using ImageJ software (National Institutes of Health, Bethesda, MD). Values are presented as arbitrary densitometry units.
Oligomeric Aβ Size Exclusion Chromatography and ELISA
The TBS-soluble fraction (500 μL) was separated according to molecular size using size exclusion chromatography on Superdex 75 10/300 GL column (GE Healthcare, Buckinghamshire, UK) in 50 mmol/L ammonium acetate pH 8.5. An AKTA fast protein liquid chromatograph (GE Healthcare) was used to collect 36 aliquots of 500 μL (28) that were used to measure Aβ by 2-site sandwich ELISA (BNT77-BA27 ELISA kit, Wako Chemicals, Richmond, VA). Molecular weight calibration was conducted using gel filtration calibration kits (GE Healthcare); the monomeric size (4 kd) was determined using native synthetic Aβ 1-40.
Statistical Analyses
Two-way analyses of variance were carried out to examine the main effects and possible interactions of Tg status and age on performances on the Morris water maze test and contextual fear conditioning (CFC), number of neurons, GFAP-positive astrocytes, amyloid plaque burden, and oligomeric Aβ load. One-way analysis of variance and Student t-test were used to test for single effects. Spearman rank test was used for correlation analyses. In all tests, the level of significance was p < 0.05. Data are presented as mean ± SEM, unless otherwise indicated.
Results
Brain Oligomeric Aβ Accumulation Is an Early and Age-Dependent Event in Tg APPsw-tauvlw Mice
Dot Blot analyses using Nab61 antibody detected soluble oligomeric Aβ in brain extracts from APPsw-tauvlw mice starting at 5 months of age (n = 12); minimal or no signal was detected in age-matched wt littermate controls (n = 12) (Fig. 1A). These results are in agreement with previous studies showing that soluble oligomeric Aβ first appears in the Tg2576 APP parental line at approximately 6 months of age (29). Size exclusion chromatography analysis and ELISA further confirmed the presence of soluble oligomeric Aβ forms in APPsw-tauvlw mice at 5 months of age, predating fibrillar Aβ deposition (amyloid plaques), astroglial cell proliferation, neuronal cell loss, and impaired cognition.
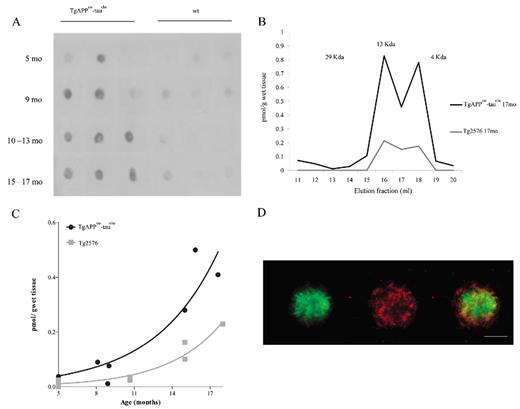
Oligomeric β-amyloid (Aβ) in brains of APPsw-tauvlw mice. (A) Dot blot analyses using Nab61, a conformation-specific antibody for oligomeric Aβ, detects oligomeric Aβ in brain extracts from APPsw-tauvlw mice starting at 5 months of age. Minimal or no signal was detected in age-matched wild-type (wt) littermate controls (n = 12 per group). (B) The molecular size of the oligomers detected by enzyme-linked immunosorbent assay and determined by size exclusion chromatography. The elution profile reveals that a large portion of the mixture consists of low-molecular-weight (LMW) oligomers, such as dimers and trimers corresponding with fractions 15-18 (8 kd-12 kd). (C) The amount of LMW oligomers correlates and exponentially increases with age (p = 0.0003). The amount of oligomeric Aβ was significantly higher in APPsw-tauvlw mice than in similarly aged mice from the parental APP line Tg2576 (n = 9 per group) (p = 0.0097). (D) Representative photomicrograph of a plaque stained with ThioS (green) and Nab61 (red). Nab61 signal is present in the dense core and in a halo surrounding the core. Scale bars = 50 μm.
The elution profile from the size exclusion chromatography column revealed that the soluble Aβ oligomeric forms detected by the ELISA were low-molecular-weight Aβ oligomers, specifically dimers and trimers, corresponding to fractions 15-18 (8 kd-12 kd) (Fig. 1B). We did not detect higher molecular weight (HMW) Aβ oligomers. This could be caused by at least 2 different possibilities: either the amount of HMW is too small to be detected by the Aβ ELISA (<0.1 pmol/g wet tissue) and/or binding molecules could mask the epitope of Aβ antibodies and interfere with HMW oligomer detection.
The amount of low-molecular-weight Aβ oligomers significantly correlated and exponentially increased with age in APPsw-tauvlw mice (p = 0.0003) (Fig. 1C). The amount of soluble oligomeric Aβ was significantly higher in the APPsw-tauvlw mice than in age-matched mice from the parental APP line Tg2576 (n = 9) (p = 0.0097) (Fig. 1C). This result is in agreement with our previous observation that APPsw-tauvlw mice accumulate in their brains larger amounts of Aβ deposits than their parental APP line Tg2576 (21).
By immunohistochemistry, oligomeric Aβ labeling with Nab61 antibody was first detected in the EC and CA1 at around 9 months of age and was clearly present in the dense core of some, but not all, amyloid plaques and in a halo surrounding the core, as described by others (30) (Fig. 1D). The amount of Nab61-reactive oligomeric Aβ deposits increased in an age-dependent fashion (p = 0.0084). No Nab61 immunostaining was detected in age- and sex-matched wt mice (not shown). Increasing amounts of sodium dodecyl sulfate-soluble abnormally hyperphosphorylated tau (Ser-202 phospho-tau) were detected by Dot Blot starting at 5 months of age in APPsw-tauvlw mice (not shown). The presence of abnormally hyperphosphorylated tau in the cytoplasm of otherwise healthy looking neurons was detected by immunostaining with CP13 antibody at around 9 months following a regional pattern of distribution that overlapped that of fibrillar amyloid plaques targeting EC, CA1, and cingulate cortex, as previously reported (31). No NFT-like formation was observed through 17 months of age in these mice.
The Amount of Oligomeric Aβ Correlated With Other Markers of Aβ Deposition but Exhibited a Differential Pattern of Regional Distribution in APPsw-tauvlw Mice
Classic 4G8-positive amyloid plaques first develop in the brains of APPsw-tauvlw mice at approximately 9 months of age in the EC, CA1 hippocampal subfield, and cingulate cortex, 3 areas known to be selectively affected in AD; their amounts increase as the mice age (21). A small subset of those amyloid deposits correspond to fibrillar dense-core ThioS-reactive plaques that are first seen at 9 months and follow an identical anatomical distribution, becoming increasingly abundant with age. Immunostaining with Nab61 labeled most, but not all, of the ThioS-reactive plaque subpopulation; ThioS-nonreactive plaques were only infrequently Nab61 positive (not shown). As expected, the total amount of brain oligomeric Aβ deposits in APPsw-tauvlw mice significantly correlated with total 4G8 immunostained amyloid burden (p < 0.0001) and ThioS-positive fibrillar dense-core plaque load (p = 0.032) (Fig. 2A-C), suggesting that levels of oligomeric Aβ correlated with these other markers of Aβ deposition. An intriguing finding, however, was that Nab61 oligomeric Aβ deposits were first detected in the EC and CA1 of APPsw-tauvlw mice at 9 months, whereas the cingulate cortex remained free of oligomeric Aβ deposits up to 13 months of age despite the presence of abundant 4G8-immunostained and ThioS-positive plaques (Fig. 3A-F). This differential pattern of distribution of classic fibrillar Aβ plaques (EC, CA1, and cingulate) and oligomeric Aβ deposits (EC, CA1, but not cingulate) at an early age in these mice may account in part for the highly selective vulnerability of the neuronal populations in medial temporal lobe structures and the relative preservation of neurons in the cingulate cortex observed in these mice.
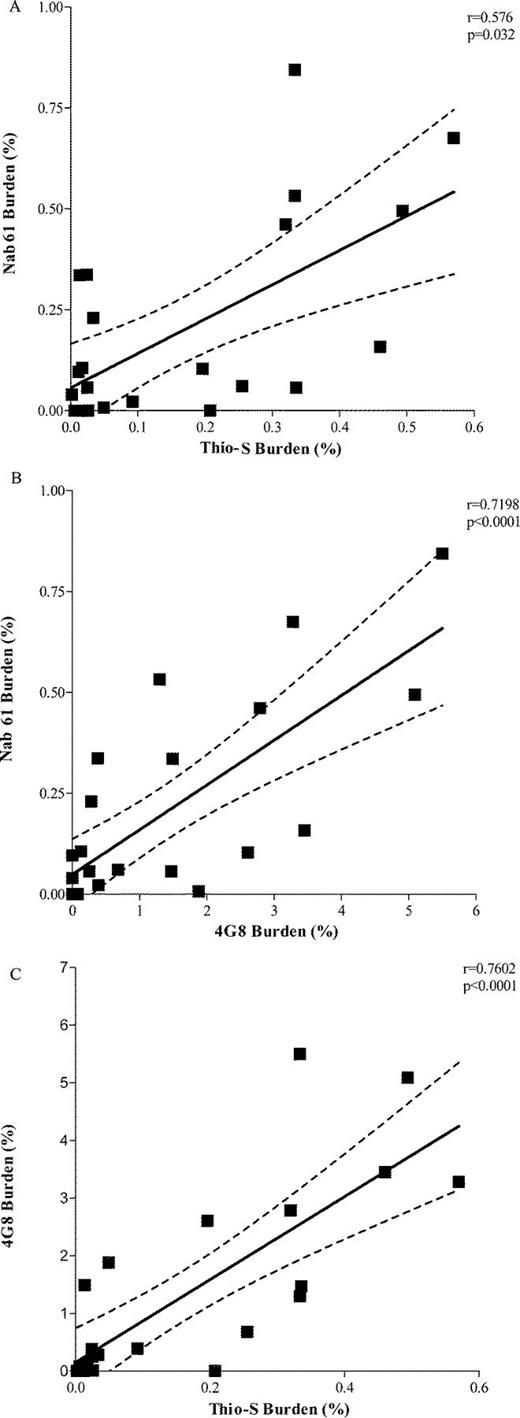
(A-C) The amount of brain oligomeric β-amyloid (Aβ deposits (Nab61 burden) in APPsw-tauvlw mice significantly correlated with thioflavin S (ThioS)-positive fibrillar dense-core plaque load (p = 0.032) (A) and with 4G8-immunostained total amyloid burden (p < 0.0001) (B). (C) 4G8 immunostained total amyloid burden significantly correlated with ThioS-positive fibrillar dense-core plaque load (p < 0.0001) (aged 5-17 months, n = 26).
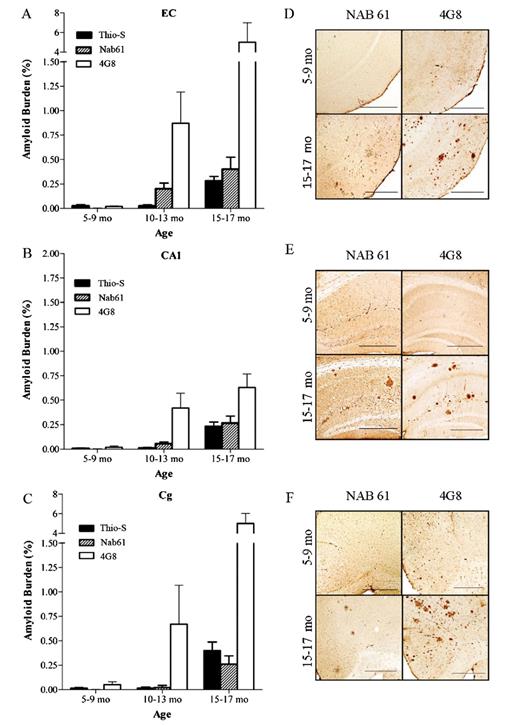
(A-C) Nab61-positive oligomeric β-amyloid (Aβ) deposits are first detected in the entorhinal cortex (EC) and the CA1 hippocampal subfield of APPsw-tauvlw mice at age 9 months, whereas the cingulate cortex (Cg) remains free of oligomeric Aβ deposits up to 13 months of age, despite very abundant amyloid plaque deposition (n = 26). (D-F) Amyloid plaques immunostained with 4G8 antibody and oligomeric Aβ deposits labeled with Nab61 antibody in EC, CA1, and Cg. Scale bars = 500 μm.
Oligomeric Aβ Deposits and Neuritic Plaques but Not Total Aβ Plaque Burden Correlated With Neuronal Cell Loss in APPsw-tauvlw Mice
We previously showed that at 9 months of age, APPsw-tauvlw mice begin to exhibit neuronal cell death in the EC, predating robust amyloid plaque deposition or NFT-like formation (21). The neuronal cell loss becomes increasingly more pronounced in the EC and extends to the CA1 subfield of the hippocampus by 12 months, along with overt classic amyloid plaque deposition, abnormal tau phosphorylation (but not NFT-like formation), and proliferation of astrocytes and microglial cells (31). By 16 months of age, neuronal loss in the EC and the CA1 hippocampal subfield approaches 40% compared with similarly aged wt littermate controls. Amyloid pathology and glial cell proliferation become very robust throughout cortical and limbic areas, and overt memory deficits can be consistently demonstrated by a hippocampal paradigm such as the Morris water maze test (21, 31).
Our current data confirmed that neuronal loss followed a highly selective pattern of regional vulnerability targeting the neuronal populations in EC and the CA1 hippocampal subfield at early ages (Fig. 4A-F). Interestingly, no significant neuronal loss was found in the cingulate cortex of APPsw-tauvlw mice through 17 months of age, despite comparable or even higher amounts of 4G8 immunostained and ThioS-reactive amyloid plaques compared with those seen in the EC and CA1 (Fig. 4A-F).
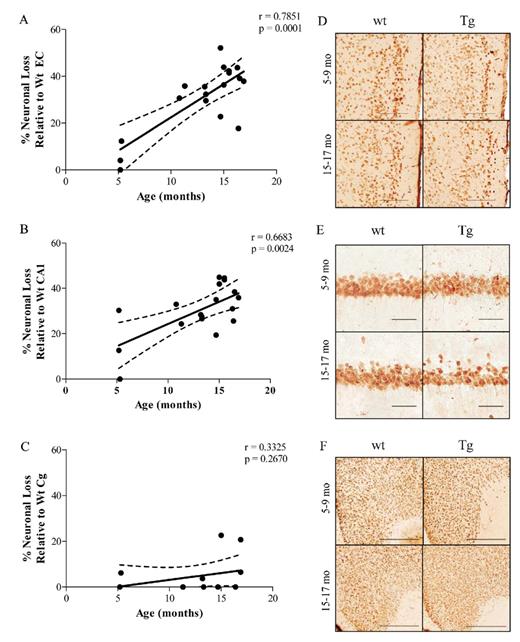
Neuronal loss in APPsw-tauvlw (Tg) mice. (A, B) Neuron loss followed a highly selective pattern of regional vulnerability targeting the populations in entorhinal cortex (EC) and the CA1 hippocampal subfield and increased with age in comparison to age- and sex-matched wild-type (wt) littermate controls. (C) No significant neuronal loss was found in the cingulate cortex (Cg) of Tg mice through 17 months of age versus age- and sex-matched wt littermate controls (p = 0.2670) (n = 18-20 per group). (D-F)Sections from wt and Tg mice stained with NeuN show neuronal loss in EC (D) and CA1 (E) and preservation of neurons in Cg (F). Scale bars = (D) 200 μm; (E) 50 μm; and (F) 500 μm. * p < 0.05, ** p < 0.01, *** p < 0.001.
The time course regional appearance of Nab61 oligomeric Aβ deposits in the brains of APPsw-tauvlw mice (EC and CA1 at 9 months, cingulate at 13 months) matched the selective pattern of regional vulnerability to neuron death, that is, neuronal populations in EC and the CA1 hippocampal subfield were lost at early ages, whereas those in the cingulate cortex through 17 months of age were spared. There were significant correlations between the amount of oligomeric Aβ deposits in the EC and CA1 and number of neurons lost in the EC (p = 0.0371) and CA1 (p = 0.0011) areas (Fig. 5A, B). There was also a significant correlation between the subpopulation of “neuritic” ThioS-reactive plaques and neuronal death in the EC (p = 0.0490) and CA1 (p = 0.0071) (Fig. 5C, D). This subpopulation of dense-core plaques (which overall represents ∼10% of total plaque burden in the APPsw-tauvlw mice) is typically surrounded by clusters of activated glial cells and strongly associated with severe dendritic and axonal changes both in AD brains and Tg mouse models (32-34). There was no correlation between total immunostained amyloid plaque burden (4G8) and numbers of neurons in the EC (p = 0.4251) or CA1 (p = 0.2571) (Fig. 5E, F). Together, these data suggest that oligomeric Aβ deposits and the subpopulation of dense-core ThioS-reactive plaques but not total Aβ plaque deposition likely contribute to neuronal damage in this model.
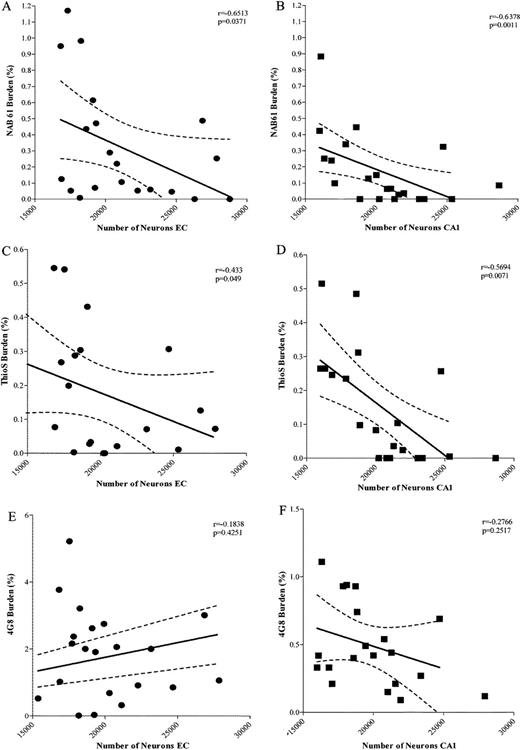
(A, B) Significant inverse correlations between the amount of oligomeric β-amyloid (Aβ) deposits and numbers of neurons in the entorhinal cortex (EC) (A) (p = 0.0371) and CA1 (B) (p = 0.0011). (C, D) There was also a significant inverse correlation between the amount of “neuritic” plaques labeled with thioflavin S (ThioS) and number of neurons in the EC (p = 0.0490) and CA1 (p = 0.0071). (E, F) There was no correlation between total amount of amyloid plaques labeled by 4G8 and numbers of neurons in EC (p = 0.4251) and CA1 (p = 0.2517) (aged 5-17 months) (n = 22).
Oligomeric Aβ Deposits and Neuritic Plaques but Not Total Aβ Plaque Burden Correlated With the Number of Reactive Astrocytes in APPsw-tauvlw Mice
The amount of oligomeric Aβ deposits (Fig. 6A, B) and ThioS-reactive plaque subpopulation (Fig. 6C, D), but not total immunostained plaque burden (Fig. 6E, F), were found to be significantly correlated with the number of GFAP-positive astrocytes in the EC and CA1 (EC p = 0.0256 and CA1 p = 0.0004; EC p = 0.0406 and CA1 p = 0.0002, respectively). This supports the idea that oligomeric Aβ forms correlate much better than total Aβ plaque burden with astrocyte inflammatory response in these mice. The numbers of GFAP-positive astrocytes were also significantly correlated with the amount of neuronal loss in the same areas (EC p = 0.0021; CA1 p = 0.0004) (Fig. 7A, B). Intriguingly, the number of GFAP-positive astrocytes in the cingulate cortex was much lower (by about 1 order of magnitude) compared with the EC and CA1 hippocampal subfield, not only in Tg mice but also in wt littermate controls at all ages. This resulted in a very large difference in the GFAP-positive astrocyte to neuron ratio among regions (EC 1:3 in wt and 1:1 in Tg mice; CA1 1:1 in wt and 2:1 in Tg mice; cingulate cortex 1:50 in wt and 1:6 in Tg mice) (Fig. 8A-F). Moreover, immunostaining with an antibody against the p50 subunit of NF-κB, a known key modulator of inflammation, revealed abundant signal mainly associated with ThioS-positive plaques in the EC and CA1 region of Tg APPsw-tauvlw mice; minimal or no signal was detected around plaques in the cingulate cortex (Fig. 9A-C). Double immunohistochemistry experiments demonstrated the colocalization of NF-κB-positive cells and GFAP-positive astrocytes (not shown). These data indicate that the inflammatory response of astrocytes targets the same brain areas that undergo early oligomeric Aβ accumulation and selective neuronal loss, raising the possibility that the activation of the NF-κB pathway in astrocytes may contribute to neuronal death in this model.
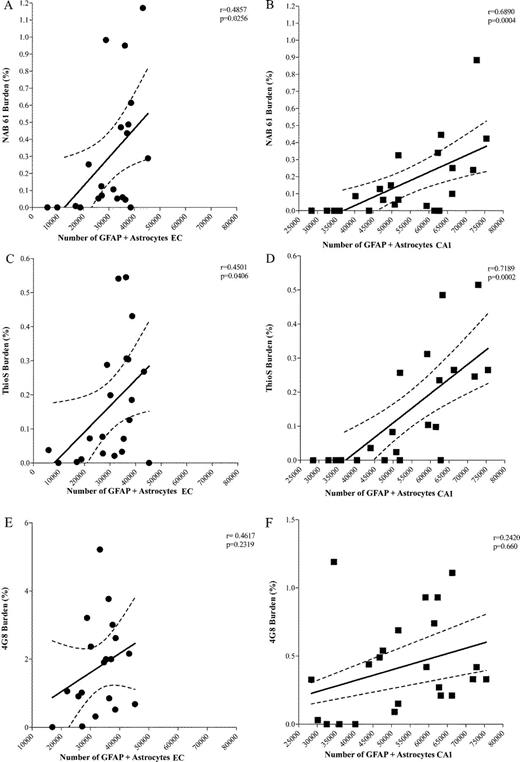
(A, B) Significant correlations between the amount of oligomeric β-amyloid (Aβ) deposits and glial fibrillary acidic protein (GFAP)-positive astrocytes in entorhinal cortex (EC) (p = 0.0256) and CA1 (p = 0.0004). (C, D) Significant correlations between the subpopulation of “neuritic” plaques labeled with thioflavin S (ThioS) and number of GFAP-labeled astrocytes in EC (p= 0.0406) and CA1 (EC p = 0.0002). (E, F) No significant correlation between total amount of amyloid plaques labeled by 4G8 and numbers of GFAP-labeled astrocytes in EC (p = 0.2319) and CA1 (p = 0.660) (aged 5-17 months, n = 22).
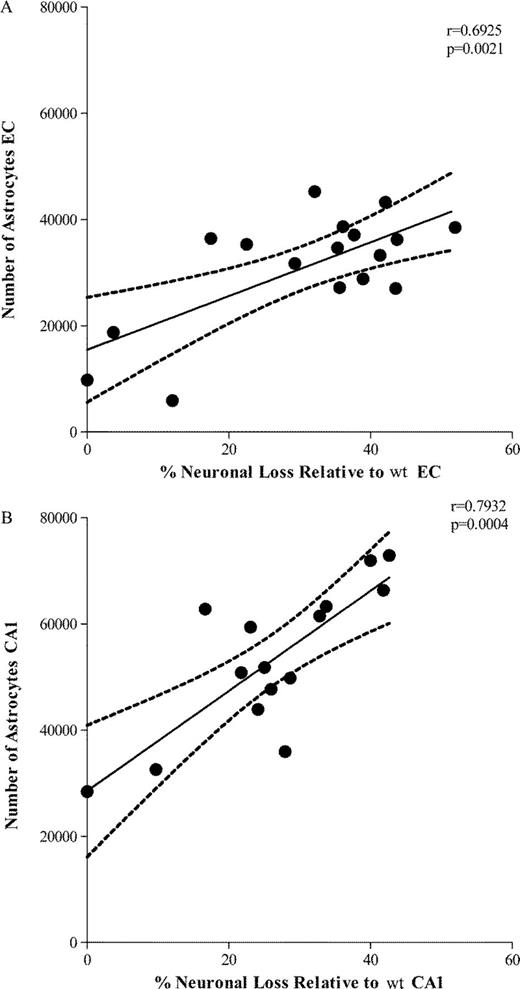
(A, B) The number of glial fibrillary acidic protein (GFAP)-labeled astrocytes significantly correlated with the amount of neuronal loss in entorhinal cortex (EC) (p = 0.0021) and CA1 (p = 0.0004). wt = wild-type.
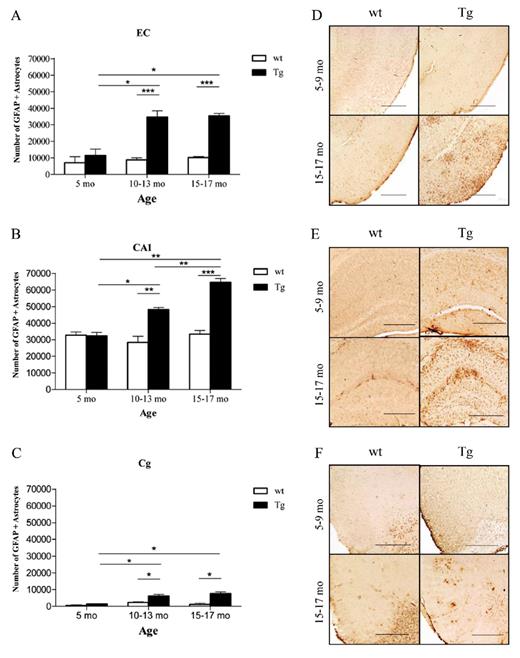
(A-C) The number of glial fibrillary acidic protein (GFAP)-labeled astrocytes was significantly higher in APPsw-tauvlw (Tg) mice compared with similarly aged wild-type (wt) littermate controls in the 3 regions examined and increased with age. The absolute number of GFAP-positive astrocytes in the cingulate cortex (Cg) was much lower by approximately an order of magnitude versus that in the entorhinal cortex (EC) and CA1 hippocampal subfield across the ages studied (5-17 months). (D-F) Representative photomicrographs of GFAP-positive astrocytes in EC (D), CA1 (E), and Cg (F) sections from wt and Tg mice. The GFAP signal in the Cg was mainly confined to the periphery of amyloid deposits, whereas there was more extensive and diffuse proliferation of astrocytes, in addition to those closely related to plaques, in EC and CA1. Scale bars = (D-F) 500 μm. * p < 0.05, ** p < 0.01, *** p < 0.001.
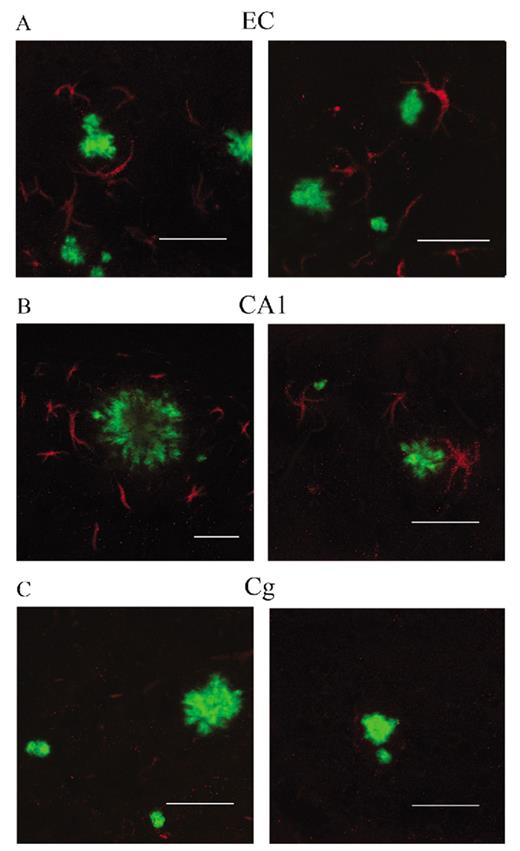
(A-C) Immunostaining with an antibody against the p50 subunit of NF-κB (red) revealed abundant NF-κB signal mainly associated with astrocytes surrounding thioflavin S (ThioS)-positive core plaques (green) in entorhinal cortex (EC) (A) and CA1 (B) of APPsw-tauvlw (Tg) mice. Minimal or no signal was detected in astrocytes around ThioS-positive plaques in the cingulate cortex (Cg) (C). Scale bars = (A-C) 50 μm.
The Number of GFAP-Positive Astrocytes Was the Best Correlate of Cognitive Performance in APPsw-tauvlw Mice
We previously showed 9 months old APPsw-tauvlw mice do not exhibit significant learning and memory deficits (as measured by the Morris water maze test) despite incipient amyloid deposition, tau phosphorylation, and neuronal loss in the EC, whereas by 16 months, they exhibit marked spatial reference memory impairment compared with wt controls (21). We also found that the total amount of 4G8-labeled amyloid plaques do not correlate well with memory impairment in these mice (21, 31). Here, we confirmed these observations and showed that there was no significant correlation either between ThioS-reactive plaques or oligomeric Aβ load and cognitive performance, as assessed by the Morris water maze and CFC paradigms (Fig. 10A-C). Although neuronal loss in the CA1 significantly correlated with freezing 24 hours after CFC training (p = 0.0331), the number of GFAP-positive astrocytes was the closest correlate of cognitive performance. The number of GFAP-positive astrocytes in the EC increased in parallel with escape latency during the invisible platform training and inversely correlated with the percent of time spent in the correct quadrant of the pool during the probe trials (r = −0.61, p = 0.0071) (Fig. 10D, E). In addition, the number of GFAP-positive astrocytes in CA1 inversely correlated with the percentage of freezing after 24 hours in the CFC paradigm (r = −0.68, p = 0.0256).
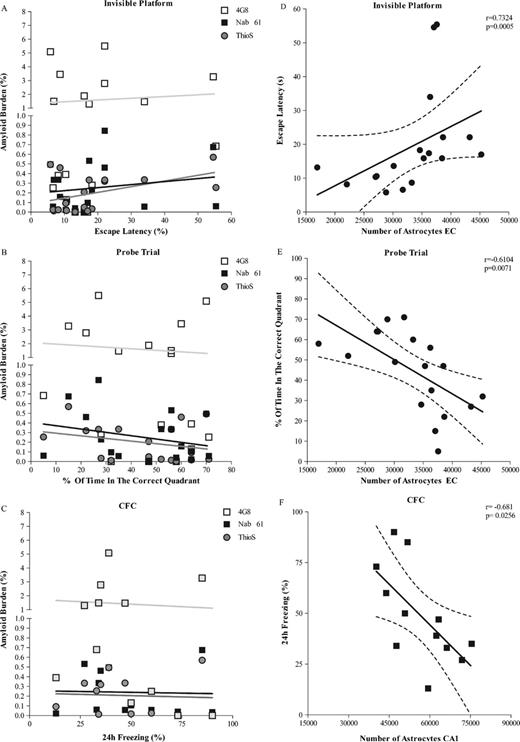
(A-C) There was no significant correlation between total amount of amyloid plaques (4G8), thioflavin S (ThioS)-reactive plaques or oligomeric β-amyloid (Aβ) load (Nab61) and cognitive performance, as assessed by the average escape latency during the invisible platform training and the percent of time in the correct quadrant of the pool during probe trials (Morris water maze test) or the percentage of freezing after 24 hours in the contextual fear conditioning (CFC) paradigm (p > 0.05). (D-F) The number of astrocytes in the entorhinal cortex (EC) (D) significantly correlated with escape latency during invisible platform training (p = 0.0005) and percent of time in the correct quadrant of the pool during probe trials (p = 0.0071) (E) (n = 18). (F) The number of astrocytes in CA1 significantly correlated with the percentage of freezing after 24 hours in the CFC paradigm (p = 0.0256) (n = 12).
Discussion
Brain accumulation of Aβ is thought to be central to AD, and it is likely to be responsible for the neurodegeneration that underlies dementia; however, the mechanistic role of Aβ and the form in which it is toxic remain controversial. Support for the hypothesis that classic fibrillar amyloid plaques are deleterious to the brain comes from numerous studies, showing that the subpopulation of dense-core Aβ plaques in particular, the so-called neuritic plaques, are intimately associated with local dendritic spine loss, changes in neurites, and gliosis in AD and mouse models (35-37). On the other hand, total numbers of amyloid plaques do not correlate well with the severity of illness (38) or with loss of neurons (3), arguing against a direct causal effect of plaques on cognition or neuronal cell death in AD. A second hypothesis that has increasing support centers on the idea that the toxic component is within the soluble fraction, more specifically that soluble Aβ oligomers cause the damage and are responsible for the toxicity. Data showing that soluble forms of Aβ correlate more closely with dementia severity than fibrillar Aβ (9, 11) and that Aβ oligomers alter dendritic spine density and affect hippocampal synaptic plasticity in vivo support this alternate hypothesis (12-14, 16, 19, 28, 39-41).
We tested these competing, although not necessarily exclusive, hypotheses by measuring brain Aβ oligomers and amyloid plaques and evaluated the form of Aβ associated with cognitive dysfunction and neuropathologic hallmarks of AD in APPsw-tauvlw mice. We found that brain accumulation of soluble small oligomeric species of Aβ (dimers and trimers) is an early event that predates by months the classic fibrillar amyloid plaque deposition, neuronal cell loss, and memory impairment, suggesting that accumulation of soluble Aβ oligomers may act as the primary trigger event in the cascade leading to neurodegeneration and impaired cognition. Interestingly, in rats, the 2-molecule soluble form of the peptide (dimers) directly isolated from AD brains is the smallest synaptotoxic species of soluble oligomeric Aβ, and it inhibits long-term potentiation (17). More recently, it has also been shown that Aβ dimers in the blood and cortical extracts from AD patients strongly correlate with cognitive impairment (10, 42). The oligomeric conformational epitope-specific antibody Nab61 immunolabeled a subset of plaques in the brains of APPsw-tauvlw mice and revealed an anatomical distribution of oligomeric Aβ deposits that partially overlapped but did not completely match that of classic amyloid plaques. Oligomeric Aβ deposits selectively appeared in the EC and CA1 hippocampal subfield of APPsw-tauvlw mice at 9 months, but only became consistently present and abundant at a substantially later age in the cingulate cortex. Because identical time course of appearance and comparable or even higher amounts of 4G8-immunostained amyloid plaques and dense-core ThioS-reactive plaques were observed in the cingulate cortex when compared with those in the EC and CA1, this was not an artifact of APP and tau transgene expression (43, 44) or the result of increased Aβ. Thus, it is possible that specific mechanisms responsible for Aβ oligomerization and/or clearance of Aβ oligomers may differ across different brain areas in these mice.
One of our most interesting observations is that the appearance of oligomeric Aβ deposits (i.e. EC and CA1 early and cingulate cortex later) faithfully predicted and matched the selective regional vulnerability of the EC and CA1 to neuronal cell loss and the resilience of neurons in the cingulate cortex through 17 months, which supports the idea that oligomeric Aβ plays a larger role than amyloid plaques in neuronal damage in this model. Further support comes from the observation that oligomeric Aβ load and the subpopulation of dense-core ThioS-reactive Aβ deposits, but not total amyloid plaque burden, correlated with the amount of neuronal cell loss.
In AD, neuronal loss is to a great extent limited to specific areas of the neocortex, limbic systems, and subcortical ascending projection systems. We reported that even at prodromal clinical stages, AD patients had a 60% of neuron loss in layer II of the entorhinal cortex and 40% loss in layer IV, as compared with controls (45). Significant neuronal loss has also been reported in AD brains in the CA1 hippocampal subfield (48%) at slightly later clinical stages (46). Our stereological analyses showed a strikingly similar regional pattern of neuronal vulnerability in the brains of APPsw-tauvlw mice, with neuronal cell death starting in the EC at 9 months and extending to CA1 at around 12 months but no significant loss of neurons in the cingulate cortex through 17 months (21, 31). Interestingly, no overt neuronal loss has been reported in the human cingulate cortex even at advanced stages of the disease, despite an observed early reduction of blood flow and metabolism on single-photon emission computed tomography and fluorodeoxyglucose-positron emission tomography in this area (47, 48). Therefore, the cingulate cortex may provide a window for studying events linked to neuronal cell death that medial temporal lobe structures (i.e. EC and CA1) pass through decades before death in humans (49) and several months in APPsw-tauvlw mice. Our present results indicate that oligomeric Aβ accumulation and activation of astrocytes are the 2 events that most faithfully predict and more closely correlate with the highly selective regional pattern of neuronal vulnerability observed in APPsw-tauvlw mice.
Inflammatory changes, including the presence of abundant activated microglia and astrocytes, and release of proinflammatory mediators are consistently identified in association with dense-core Aβ plaques from early stages of AD and in Tg mouse models, pointing to inflammation as an important pathogenetic component of the disease. Interestingly, in vitro studies have shown differential effects of oligomeric and fibrillar Aβ on astrocyte-mediated inflammation (50). Oligomeric Aβ induced a profound early inflammatory response, whereas fibrillar Aβ showed less increase of proinflammatory molecules (i.e. interleukin-1β, tumor necrosis factor, and NO), consistent with a more chronic form of inflammation. In the present study we noted increasing amounts of GFAP-positive astrocytes in the EC and CA1 of APPsw-tauvlw mice that tightly correlated with the amount of oligomeric Aβ deposits and ThioS-positive cored plaques, but not with the total amyloid plaque burden, further suggesting that oligomeric Aβ species along with the subpopulation of “neuritic” plaques likely play a larger role than total amyloid plaque deposition in the brain inflammatory glial cell response in these animals. The numbers of GFAP-positive astrocytes very closely paralleled the amount of neuronal loss in the EC and CA1. Moreover, the total number of GFAP-positive astrocytes in the cingulate cortex was much lower than in the EC and CA1, both in Tg and WT littermates. This intriguing numerical inferiority in the amount of reactive astrocytes in the cingulate cortex (which rendered a much smaller GFAP-positive astrocyte to neuron ratio than in the EC and CA1) might account, at least in part, for the comparatively less neuronal vulnerability than that observed in the medial temporal lobe structures in APPsw-tauvlw mice and the apparent relative resilience of cingulate cortex neurons. Furthermore, immunostaining with an antibody against NF-κB, a key regulator of proinflammatory genes (e.g. interleukin-1β tumor necrosis factor or cyclooxygenase 2) in glial cells, revealed abundant signal associated with astrocytes surrounding ThioS-positive cored plaques in the EC and CA1 region of Tg APPsw-tauvlw mice but minimal signal in astrocytes in the cingulate cortex. These results raise the possibility that oligomeric Aβ, alone or in addition to fibrillar Aβ in dense cored plaques, could contribute to neuronal toxicity via astrocytic NF-κB activation. Favoring this possibility, in vitro studies have shown that Aβ exposure induces NF-κB activation and NO production in astrocytes (51). Moreover, coculture experiments demonstrated that exposure of primary human astroglia to Aβ resulted in NF-κB activation in astrocytes; those Aβ-activated astrocytes induced in turn apoptotic cell death in primary human neurons (52). Moreover, Aβ exposure can cause alterations in glucose metabolism in astrocytes and this in turn impairs neuronal viability and functionality (53). Therefore, the possibility exits that oligomeric Aβ-mediated activation of NF-κB in astrocytes may contribute to neuronal cell death in AD. This hypothesis needs now to be further investigated, as it might prove relevant for therapeutic intervention.
Despite the tight correlations observed between oligomeric Aβ load and number of neurons lost and GFAP-positive astrocytes in the brains of APPsw-tauvlw mice, neither oligomeric Aβ load nor other morphological markers of Aβ deposition correlated well with memory performance. The number of GFAP-positive astrocytes in EC and CA1 hippocampal subfield was by far the parameter most intimately correlated with the severity of spatial reference and contextual memory impairment in these mice. Based on these data, we suggest that proliferation and activation of astrocytes is very likely a driving force for impaired cognition in this model. This is consistent with our recent data showing that oral chronic treatment with Triflusal, an agent with potent anti-inflammatory effects in the CNS via inhibition of NF-κB activation, significantly decreased the number of reactive astrocytes and microglial cells and rescued memory deficits in Tg2576 mice despite its lack of impact on total amyloid plaque deposition (54).
In conclusion, the present work shows that oligomeric Aβ, but not total amyloid plaque accumulation, in the brain of Tg APPsw-tauvlw mice is an early event that predates and faithfully predicts the selective pattern of regional neuronal vulnerability and closely correlates with the extent of neuronal cell loss. Oligomeric Aβ and fibrillar Aβ in dense-core plaques, but not total diffuse plaque burden, are intimately associated with astrocytic proliferation in the brains of these mice. The number of GFAP-positive astrocytes is by far the parameter most tightly correlated with memory impairment and neuronal cell death in these mice, and astrocyte immunoreactivity to NF-κB is markedly increased in the same medial temporal lobe regions that undergo early oligomeric Aβ accumulation and selective neuronal degeneration. Our observations place astrocytes as a target of oligomeric Aβ and support the hypothesis that the astrocyte inflammatory response, likely mediated by an NF-κB-related mechanism, is a driving force for impaired cognition and may also contribute to neuronal cell damage. Further studies are now needed to demonstrate a causal relation that might prove relevant in AD.
Acknowledgment
The authors thank Dr. Virginia Lee for kindly providing the Nab61 antibody.
References
Author notes
This project was funded in part by FIS PI041887 and CIBERNED.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}