





Abstract
Tissue hypoxia and necrosis represent pathophysiologic and histologic hallmarks of glioblastoma (GBM). Although hypoxia inducible factor 1α (HIF-1α) plays crucial roles in the malignant phenotypes of GBM, developing HIF-1α-targeted agents has been hampered by the lack of a suitable preclinical model that recapitulates the complex biology of clinical GBM. We present a new GBM model, MGG123, which was established from a recurrent human GBM. Orthotopic xenografting of stem-like MGG123 cells reproducibly generated lethal tumors that were characterized by foci of palisading necrosis, hypervascularity, and robust stem cell marker expression. Perinecrotic neoplastic cells distinctively express HIF-1α and are proliferative in both xenografts and the patient tissue. The xenografts contain scattered hypoxic foci that were consistently greater than 50 μm distant from blood vessels, indicating intratumoral heterogeneity of oxygenation. Hypoxia enhanced HIF-1α expression in cultured MGG123 cells, which was abrogated by the HIF-1α inhibitors digoxin or ouabain. In vivo, treatment of orthotopic MGG123 xenografts with digoxin decreased HIF-1α expression, vascular endothelial growth factor mRNA levels, and CD34-positive vasculature within the tumors, and extended survival of mice bearing the aggressive MGG123 GBM. This preclinical tumor model faithfully recapitulates the GBM-relevant hypoxic microenvironment and stemness and is a suitable platform for studying disease biology and developing hypoxia-targeted agents.
Introduction
Glioblastoma (GBM) is the most common human malignant primary brain tumor and accounts for 60% to 70% of malignant gliomas. The incidence of GBM is estimated to be approximately 3.2/100,000 per year and carries a prevalence of 28/100,000 per year ( 1 ). Current prognosis of GBM remains dismal and has not improved substantially over the last few decades, with a 5-year survival rate below 5%; most patients die within 2 years after diagnosis ( 2 ).
Hypoxic regions are frequently found in GBM and result in extensive necrosis that is a histologic hallmark of World Health Organization (WHO) grade IV GBM, distinguishing it from lower-grade gliomas ( 3, 4 ). Necrotic foci in GBM are typically surrounded by viable palisading cells, a unique configuration seen in GBM; the presence of necrosis in gliomas is recognized as a negative prognostic marker ( 5–7 ). Accumulating evidence supports an important role for tissue hypoxia in GBM growth, neovascularization, invasion, resistance to chemo- and radiotherapy, and ultimately recurrence after treatment ( 8–14 ). Hypoxia is considered to promote fundamental molecular and phenotypic changes related to malignant progression such as loss of apoptotic potential, oncogene activation, gene amplifications, and genetic instability ( 13, 15–17 ). Furthermore, hypoxia also provides a niche microenvironment to maintain brain tumor stem cells ( 18–22 ).
The hypoxia-inducible factor (HIF) family of transcriptional factors is a master regulator of diverse cellular responses to hypoxia ( 12, 14, 23 ). In humans, 3 HIFα subunits (i.e. HIF-1α, HIF-2α, and HIF-3α), which are all oxygen level sensitive, have been identified. Among them, HIF-1α is the most ubiquitously expressed and induced in cells at low oxygen levels ( 24 ). Palisading GBM cells surrounding an area of necrosis are exposed to severe hypoxia and overexpress HIF-1α, thereby promoting the secretion of proangiogenic factors ( 9, 25, 26 ). High-level expression of HIF-1α has been positively correlated with tumor progression and poor prognosis in patients with GBM ( 25–27 ). Small molecule or siRNA-mediated inhibition of HIF-1α can induce apoptosis in glioma cells, sensitize them to chemotherapeutics ( 28, 29 ), and impair GBM cell migration and invasion in vitro under normoxic and hypoxic conditions ( 30 ). Thus, HIF-1α plays a pivotal role in GBM survival, resistance, and invasion. HIF-2α displays a different expression pattern than HIF-1α ( 31 ) and has been shown to be expressed in GBM stem cells ( 20 ).
All the above-mentioned observations suggest that targeting the hypoxic microenvironment or interfering with the hypoxic response of tumor cells may offer an effective avenue toward targeted treatment of GBM. Developing and assessing this strategy requires preclinical models that accurately reflect the complex biology of human GBM, composed of tumor and stromal cells at varying oxygen levels. The current preclinical models, however, mostly fail to recapitulate the histopathologic hallmarks (e.g. necrosis with palisading) of human GBM ( 32, 33 ), and thus pose limitations in developing therapy specifically targeting tumor hypoxia.
We here report a new preclinical model that closely mirrors the necrosis and palisading patterns of human GBM. This model is based on stem-like GBM cells that we isolated from a recurrent GBM. Using this model, we show that HIF-1α inhibitor digoxin inhibits HIF-1α expression and angiogenesis in vivo and provides survival benefits. This new GBM model will facilitate the understanding of hypoxia-related GBM pathophysiology and allow for further preclinical studies investigating novel therapeutic agents specifically targeting the hypoxic microenvironment of GBM.
Materials and Methods
Isolation and Culture of MGG123 and Other GBM Stem-like Cells
Human GBM stem-like cell line MGG123 was isolated from an abdominal mass that had disseminated through a ventriculoperitoneal shunt inserted in a 57-year-old patient with recurrent GBM. Tissue was processed as previously described ( 34 ). Other GBM stem-like cells were established from fresh surgical specimens, as previously described ( 34, 35 ), according to the protocol approved by the institutional review board. GBM stem-like cells were cultured as spheres in EF20 stem cell medium composed of Neurobasal medium (Invitrogen, Carlsbad, CA), supplemented with 3 mmol/L of L-glutamine (Mediatech, Manassas, VA), 1X B27 supplement (Life Technologies, Rockville, MD), 0.5 X N2 supplement (Life Technologies), 2 μg/mL of heparin (Sigma-Aldrich, St. Louis, MO), 20 ng/mL of recombinant human EGF (R&D Systems Inc., Minneapolis, MN), 20 ng/mL of recombinant human FGF2 (Peprotech, Rocky Hill, NJ), and 0.5X penicillin G-streptomycin sulfate-amphotericin B complex (Mediatech) ( 34, 35 ). The cultures were fed every 3 days with one-third volume of fresh medium. Cell passaging was performed by dissociation of spheres using TrypLE Express (Invitrogen).
Orthotopic Xenografting
Seven- to eighth-week-old female SCID mice were purchased from the National Cancer Institute (Bethesda, MD). GBM stem-like cells (30,000–50,000 cells) suspended in 3 μL of media were stereotactically implanted into the brains (right striatum, 2.5 mm lateral from Bregma and 2.5 mm deep) of severe combined immunodeficiency (SCID) mice under anesthesia with intraperitoneal pentobarbital. Autopsy tissue from a GBM patient (A1) was dissociated with trypsin, and resulting cells were immediately implanted into the brains of SCID mice. Mice were monitored and euthanized when they developed significant neurologic signs.
![Orthotopic MGG123 xenografts recapitulate the histopathologic characteristics of the patient GBM. (A) Low magnification of an H&E-stained section of a mouse brain bearing a MGG123-derived intracerebral xenograft (left panel). Arrow indicates a large area of necrosis. Higher-magnification of the boxed area on the left panel showing ill-demarcated tumor brain interfaces and tumor invasiveness (right panel). (B, C) H&E staining of orthotopic MGG123 xenografts (B) and the patient tumor (C) showing necrotic foci with palisades. N, necrosis. (D–G) Immunohistochemical characterization of the MGG123 model and the original GBM tissue. Positivity is indicated by brown. Staining for human nestin (D) shows positivity in tumor cells in both xenografts [T] and the patient’s specimen. Surrounding mouse cells in the brain [B] are negative. CD44 is homogeneously positive in both tumor tissues (E) . Sox2 positivity is prominent at perinecrotic and perivascular areas in the xenografts and patient. Arrows point to blood vessels (F). CD34 staining reveals tortuous and dilated vasculature at the tumor periphery in the xenografts ( G , left panel). CD31 staining of the patient section reveals similar vasculature (g, right panel). Scale bars: a , left panel, 1 mm; all other panels, 100 μm.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jnen/74/7/10.1097_NEN.0000000000000210/3/m_jnen_710_f1.jpeg?Expires=1716598129&Signature=B0HFErR~YjITkQYUPoBl5WD3FbBDrjKsJqJnUhyNDhy5GAarenxFBu~X8SkO9ypVL90ss4rs-dZIztX3VZdH~raUqPYG026DzEfiVCYt~~taLVrHWNY2AxRAy65qfxZpsz42ZoweeeKHxVm7r4yNvNG29Bs4Gmt87rLHC1pumG8p~DBB7I0cG7vEeldsb9T5h3McTa962N8VF7WmY4IoUU2j2uKZk4LqWEsn0ATI3eVq3QegfVtBDRRH1ppSLPC4-pvI77xwIByl6udOy8IjHns-KOTtL8M49uYow5y2DgKmBDvabl5rWaWshUbDyATRxkPPxBNW-1pEi4GQ3FBGzA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Orthotopic MGG123 xenografts recapitulate the histopathologic characteristics of the patient GBM. (A) Low magnification of an H&E-stained section of a mouse brain bearing a MGG123-derived intracerebral xenograft (left panel). Arrow indicates a large area of necrosis. Higher-magnification of the boxed area on the left panel showing ill-demarcated tumor brain interfaces and tumor invasiveness (right panel). (B, C) H&E staining of orthotopic MGG123 xenografts (B) and the patient tumor (C) showing necrotic foci with palisades. N, necrosis. (D–G) Immunohistochemical characterization of the MGG123 model and the original GBM tissue. Positivity is indicated by brown. Staining for human nestin (D) shows positivity in tumor cells in both xenografts [T] and the patient’s specimen. Surrounding mouse cells in the brain [B] are negative. CD44 is homogeneously positive in both tumor tissues (E) . Sox2 positivity is prominent at perinecrotic and perivascular areas in the xenografts and patient. Arrows point to blood vessels (F). CD34 staining reveals tortuous and dilated vasculature at the tumor periphery in the xenografts ( G , left panel). CD31 staining of the patient section reveals similar vasculature (g, right panel). Scale bars: a , left panel, 1 mm; all other panels, 100 μm.
Immunohistochemistry
Formalin-fixed tumor samples were embedded in paraffin and 7-μm-thick sections were obtained. Hematoxylin and eosin (H&E) staining was performed using standard procedures. Immunohistochemistry (IHC) was performed using antigen retrieval with microwave treatment in 1 mmol/L EDTA (pH 8, for CD31) or 10 mmol/L sodium citrate buffer (pH 6, for all the other antibodies) and Vectastain elite kit (Vector Laboratories, Burlingame, CA), as described previously ( 34, 35 ). Primary antibodies used include anti-HIF-1α (Santa Cruz Biotechnology, Santa Cruz, CA), anti-CD34 (Abcam, Cambridge, MA), anti-CD31 (Dako, Carpinteria, CA), anti-nestin (Santa Cruz Biotechnology), anti-CD44 (Cell Signaling Technology, Beverly, MA), anti-Sox2 (R&D Systems), and anti-Ki-67 (MIB-1, Dako). Signals were visualized with diaminobenzidine (Dako). Hematoxylin was used to counterstain nuclei. All specimens were examined under a microscope (Nikon) equipped with a digital camera (RT Color SPOT) connected to SPOT imaging software (Diagnostic Instruments Inc., Sterling Heights, MI).
Flow Cytometry
Cells were stained with APC-anti-CD133 (Miltenyi Biotech, San Diego, CA) and FITC-anti-CD44 (eBioscience, San Diego, CA) antibodies or APC or FITC-isotype controls according to the manufacturer’s recommendations. Cells were postfixed in 2% paraformaldehyde and analyzed by a FACSCalibur (BD Biosciences, San Jose, CA). Data were analyzed using FlowJo software (Tree Star, Ashland, OR).
Immunofluorescence Detection of Hypoxic Areas and CD34
Immunofluorescence detection of hypoxic regions was performed as previously described ( 22 ). Briefly, SCID mice bearing 3-week-old orthotopic MGG123 xenografts were given intraperitoneal injection of 60 mg/kg Hypoxyprobe (pimonidazole hydrochloride). One hour later, mice were killed, cardiac-perfused with 4% paraformaldehyde, the brains removed, and frozen sections obtained. Slides were stained with anti-Hypoxyprobe (1:50) and anti-CD34 (Abcam, 1: 100), followed by incubation with Alexa Fluor 488 goat anti-mouse IgG and Alexa Fluor 546 goat anti-rat IgG (both from Invitrogen). Slides were then mounted with 4′,6-diamidino-2-phenylindole (DAPI)-containing media (Vector Laboratories) before microscopic observation. Images were captured using SPOT imaging software.
Western Blot
MGG123 cells were treated with dimethyl sulfoxide (DMSO), digoxin (1 μmol/L), or ouabain (1 μmol/L) and placed in an incubator under normoxia (21% O 2 ), or hypoxia, (4%), for 12 hours. Cells were collected and lysed in RIPA buffer (Boston Bioproducts, Ashland, MA) with protease inhibitors (Complete Mini; Roche Diagnostics, Mannheim, Germany). Protein concentrations were determined by modified Bradford assay (Bio-Rad, Hercules, CA). Proteins (20 μg) were separated on an SDS-polyacrylamide gel, transferred to polyvinylidene difluoride membranes (Bio-Rad), and incubated with primary antibodies, anti-HIF-1α (BD Biosciences) or anti-Actin (Sigma Aldrich), at 4°C overnight. Membranes were washed and incubated with respective secondary antibodies (1:5000, anti-mouse horseradish peroxidase [HRP] or anti-rabbit HRP, Promega, Madison, WI) at room temperature for 1 hour. The proteins were visualized using the ECL plus Western blotting detection system (GE Healthcare, Little Chalfont, UK), on Kodak films.
In Vivo Studies With Digoxin Treatment
For the pharmacodynamics study, 10 SCID mice were stereotactically implanted with MGG123 cells (10 5 cells in 3 μL) into the striatum and randomized into 2 groups. One group was treated with digoxin at 2 mg/kg intraperitoneally for 5 consecutive days (from day 29 to 33); the second group received vehicle (10% phosphate-buffered saline) treatment during the same period. Three hours after the last dosing, all mice were killed and the brains were removed for IHC. HIF-1α staining was graded as – (negative), + (scattered positive cells, or 1–2 positive areas per tumor), and ++ (3 or more positive areas per tumor). The number of CD34-positive blood vessels was counted on 3 randomly chosen 100× power fields per tumor, and the averages were reported as blood vessel densities.
To study survival, 10 5 or 2.5 × 10 4 cells were implanted to generate orthotopic MGG123 xenografts in SCID mice. Treatment with digoxin (2 mg/kg, n = 6) or 10% DMSO (n = 6) started 7 days after tumor implantation and continued until mice exhibited clinical signs associated with brain tumors. Mice were monitored daily, and body weights were recorded. Mice were euthanized when they developed significant neurologic deficits or lost more than 15% of their starting weight. All animal procedures were approved by the institutional animal care and use committee.
Quantitative Reverse Transcription–Polymerase Chain Reaction
SCID mice bearing intracerebral MGG123 xenografts were treated with vehicle (n = 3) or digoxin (n = 3) for 5 consecutive days. Mice were killed, and total RNA was extracted from tumors using Trizol (Invitrogen). cDNA was synthesized with high-capacity cDNA reverse transcription kit (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA) and oligo(dT)12–18 primers (Invitrogen). cDNA and specific primers for VEGFA were then used for polymerase chain reaction (PCR) amplification using SYBR Green PCR Master Mix (Applied Biosystems) in StepOnePlus Real-Time PCR System (Applied Biosystems) followed by analysis with StepOne Software v2.3 (Applied Biosystems). GAPDH was used as housekeeping gene control. Primer sequences are GAPDH forward, CAATGACCCCTTCATTGACC; reverse, GACAAGCTTCCCGTTCTCAG; VEGFA forward, AAGGAGGAGGGCAGAATCAT; and reverse, CACACAGGATGGCTTGAAGA.
Statistical Analysis
Student t -test (2-tailed) was used to analyze differences between 2 groups. Kaplan-Meier analysis and log-rank test were used to analyze overall survival of mice receiving different treatments.
Results
Histopathologic Characterization of MGG123-Derived Orthotopic Xenografts
Intracerebral implantation of 3 × 10 5 MGG123 cells into SCID mice reproducibly generated lethal tumors. H&E stains revealed massive tumors in the implanted (right) hemispheres that displayed invasiveness and caused midline shift and severely compressed the right lateral ventricles (Fig. 1A). Notably, even under a low magnification, large necrotic areas were obvious within the tumors (Fig. 1A). Tumors also displayed invasiveness along superficial and subpial brain regions (Fig. 1A). Higher magnifications of H&E-stained sections showed densely populated atypical neoplastic cells as well as scattered necrotic foci surrounded by cells exhibiting “palisading necrosis” (Fig. 1B). These pathognomonic features of GBM were also seen in the original MGG123 tumor (Fig. 1C), indicating the phenotypic recapitulation achieved in the MGG123 model.
Immunohistochemical analysis of the xenografts demonstrated intense immunopositivity for human nestin in nearly 100% of tumor cells, clearly distinguishing neoplastic cells from host mouse cells, and phenocopying the strong nestin positivity in the original tumor (Fig. 1D). Similarly, immunostaining for CD44, a marker for a stem and mesenchymal phenotype, showed strong expression in nearly all tumor cells in both the xenografts and the patient (Fig. 1E). Another stem cell marker, Sox2, was also highly expressed, and the immune-positivity appeared prominent in perinecrotic and perivascular areas (Fig. 1F). In vitro, sphere-cultured MGG123 cells had strong expression of CD44 but lacked CD133, suggestive of a mesenchymal phenotype (Figure, Supplemental Digital Content 1, Supplementary Data ). IHC for the endothelial marker CD34 revealed aberrant dense vasculature characterized by tortuous, dilated, and sprout vessels that are mostly seen in the peripheral regions of the tumor, whereas vasculature in the unaffected brain was organized and not dilated (Figure, Supplemental Digital Content 2, Supplementary Data ). CD31 IHC on the patient tumor identified proliferation of dilated blood vessels that resembles the vasculature seen in the xenografts, and multilayered endothelial proliferation was not identified in either patient or xenografts (Fig. 1G). Thus, these analyses established the ability of the orthotopoic MGG123 model to recapitulate the histopathologic and biological characteristics of the patient GBM, including the hypoxic/necrotic tumor microenvironment, the stem-like and mesenchymal phenotype, and the morphologically abnormal vasculature, which are all hallmarks of GBM.
HIF-1α and MIB-1 Expression in the MGG123 Xenografts and Patient Tumors
Because necrosis is likely driven by tissue low oxygen tension/hypoxia within GBM, we next examined the expression of HIF-1α, which is best described as a “low oxygen sensor” in the patient tumor and the xenografts. Consistent with previous observations ( 9, 25, 26 ), HIF-1α immunopositivity was found specifically in palisading cells surrounding necrosis in the patient specimen (Fig. 2A); this distinct pattern of HIF-1α distribution was mimicked by the MGG123 xenograft (Fig. 2B). Given the role of hypoxia in stabilizing HIF-1α, this anatomical distribution of HIF-1α staining suggests that GBM contains regions with varying levels of O 2 tissue tension and that areas adjacent to necrosis are hypoxic.
![Perinecrotic neoplastic cells express HIF-1α and are proliferative in the MGG123 model and the patient GBM. (A, B) HIF-1α immunostaining shows HIF-1α expression (brown) in cells surrounding necrotic foci [N] in both the xenograft and patient tumor. (C, D) MIB-1 is highly expressed in perinecrotic areas as well as other viable tumor areas in both xenografts and patient tumor (labeling index 48% and 44%, respectively). Scale bar = 100 μm.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jnen/74/7/10.1097_NEN.0000000000000210/3/m_jnen_710_f2.jpeg?Expires=1716598129&Signature=EVjYF8VHOMB4n0QJ0oY7KVlVbqxXyHhksyCWq6ZD0sOcw6I2VPPvQ49FvtiYWvhrvZSocgPiqj2WdDzmpKyKR3sByqApbzQAtyp8hemUJ4bsDdkgOw2rmPZTINtJ6sUYSIiLmRPp4c69lqGiz77ooeEBgZ-zSm2XHW4t87pRoT0sMZtfUkBx0FA3u6-b7UFP5N8va1d2yoIOLnbCoscoWnrUOB6xyAlJX3HczKMk289lI~a4fgBLWixcvlhnvpdeWMJKjyPFipmZp6LSWf2RNAzzNwHyhyjD6uLnkDkGbC93RtsjTEjpHe8k4b7UGO0Yz4wM5NTHTcwcg~i0WDdOYA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Perinecrotic neoplastic cells express HIF-1α and are proliferative in the MGG123 model and the patient GBM. (A, B) HIF-1α immunostaining shows HIF-1α expression (brown) in cells surrounding necrotic foci [N] in both the xenograft and patient tumor. (C, D) MIB-1 is highly expressed in perinecrotic areas as well as other viable tumor areas in both xenografts and patient tumor (labeling index 48% and 44%, respectively). Scale bar = 100 μm.
We next performed MIB-1 IHC to identify actively dividing cells. Both patient and xenografted MGG123 tumors show very high MIB-1 labeling indices: 44% in the patient and 48% in the xenografts, revealing the robust proliferative activity of these tumor cells (Figs. 2C, D). Unlike the distribution pattern of HIF-1α, MIB-1 positivity was mostly homogeneous throughout the tumor, except in necrotic cores where there were no viable cells, including HIF-1α-negative areas away from necrosis and HIF-1α-positive areas surrounding necrosis. This result indicates that viable tumor cells have comparable proliferative activity irrespective of HIF-1α status. The similarities of the marker status between the xenografts and the patient’s tissue confirm that the MGG123 model accurately represents the biology and hypoxic microenvironment of human GBM.
HIF-1α and Necrosis Status in a Cohort of Patient-Derived Orthotopic GBM Xenografts
We have established a panel of highly tumorigenic stem-like cells that were isolated from patient GBMs ( 34, 35 ). Taking advantage of this resource, we screened for the presence of necrosis and the status of HIF-1α immunopositivity in a cohort of intracerebral GBM xenografts from both primary and recurrent tumors (n = 19, including MGG123). Most tumors other than MGG123 did not contain necrotic foci, whereas 2 tumors had a few small necrotic foci (Fig. 3A). Nuclear staining of HIF-1α in scattered cells was seen in 5 tumors, and a few (<3) small positive areas in 1 tumor, all of which were much less than in MGG123 tumors (Figs. 3A, B). Several tumors exhibited cytoplasmic or perinuclear HIF-1α staining in a small number of cells; the biological significance of such HIF-1α localization in cancer has been controversial ( 36–38 ). These results illustrate the unique hypoxic characteristics of the MGG123 model.
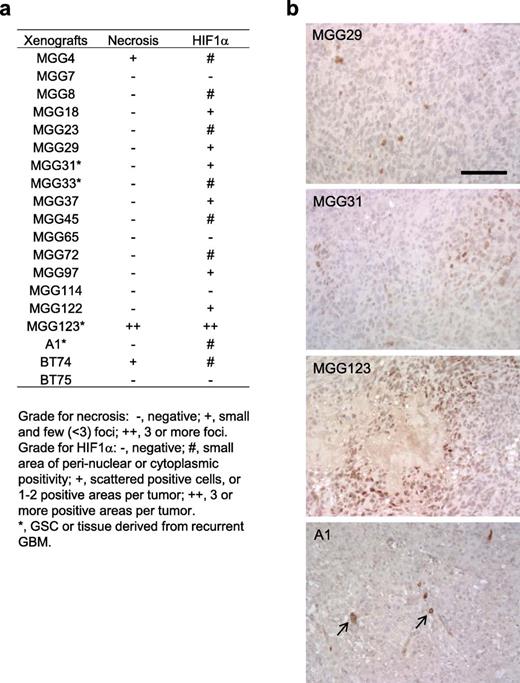
HIF-1α and necrosis status in a cohort of patient-derived orthotopic GBM xenografts. (A) Summary of the status of necrosis and HIF-1α IHC in 19 patient-derived orthotopic GBM xenografts. (B) Examples of HIF-1α IHC on orthotopic GBM xenografts. Some tumors contain a small number of cells that exhibit HIF-1α staining in the perinuclear areas or cytoplasm (arrows). Scale bar = 100 μm.
Anatomical Relationship Between Hypoxic Areas and Blood Vessels in Orthotopic MGG123 Tumors
Next, we sought to analyze the anatomical relationship between tumor-associated vasculature and hypoxic regions within the orthotopic MGG123 xenografts. Hypoxyprobe (pimonidazole) was injected to tumor-bearing mice; its product in hypoxic cells as well as the vascular endothelium marker CD34 was visualized using immunofluorescence. A number of foci of hypoxia were observed within the tumors, mostly at or near the center of the tumor (Fig. 4A), whereas hypoxic areas were not detected in the contralateral normal cerebral cortex (Fig. 4B). Measurements of the distance from each CD34-positive blood vessel to the nearest hypoxic region revealed distances ranging from 60 to 230 μm, with an average of 125 μm, and no hypoxic areas were found <50 μm from blood vessels (Fig. 4C). We also found that CD34 positivity is not present within hypoxic areas. Thus, substantial oxygenation heterogeneity exists within MGG123 tumors and the distance from blood vessels likely is a major determinant of tissue oxygen concentrations. In addition, large hypoxic foci contained an area without DAPI-stained nuclei at their cores, suggesting that hypoxic regions progress to necrosis (Fig. 4D).
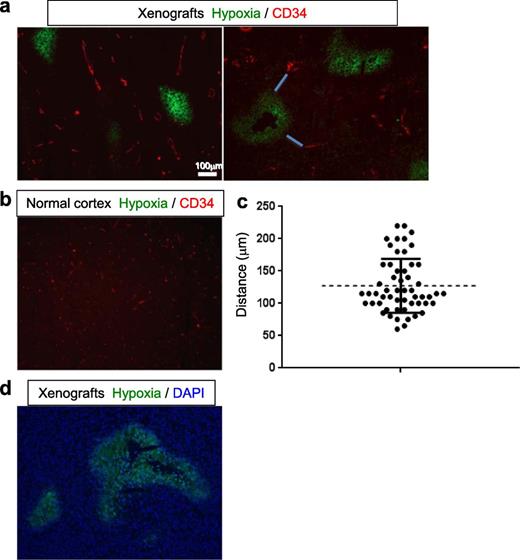
Anatomical correlation of hypoxia areas and blood vessels in the orthotopic MGG123 xenografts. (A, B) Immunofluorescence evaluation of the presence of hypoxic areas (green) and CD34-positive blood vessels (red) in orthotopic xenografts ( A , representative images from 2 animals) and the contralateral normal cerebral cortex (B) . (C) Plot showing the result of measurements of distances between CD34-positive blood vessels and the nearest hypoxic area. Examples of such distances are shown with blue lines in the right panel of (A) . (D) Merged image of hypoxia (green) and DAPI (blue) showing absence of cells at the center of a large region of hypoxia.
Hypoxia Promotes HIF-1α Expression, Which Is Blocked by Cardiac Glycosides in MGG123 Cells
We next tested whether MGG123 cells respond to hypoxic conditions by increasing HIF-1α protein levels in vitro. Western blot analysis showed that HIF-1α protein levels, which were detectable under normoxia, greatly increased after 12-hour exposure to hypoxia (4% O 2 ) (Fig. 5A).
![Cardiac glycosides suppress HIF-1α expression in vitro and in vivo. (A) Western blot showing hypoxia induction of HIF-1α in MGG123 cells and its abrogation by digoxin [D] and ouabain [O] (1 μmol/L). N, normoxia. Actin was used as a loading control. (B) IHC for HIF-1α on sections derived from an in vivo pharmacodynamic study testing systemic digoxin treatment. Upper panels show results obtained in 5 mice in the vehicle group (V1–V5); bottom panels show matching results from 5 mice in the digoxin group (D1–D5). B, brain; T, tumor. Scale bar = 100 μm. (C) Table summarizing the HIF-1α immunopositivity status in all the animals.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jnen/74/7/10.1097_NEN.0000000000000210/3/m_jnen_710_f5.jpeg?Expires=1716598129&Signature=BL0xbBhE9BmsR0yuCAmeiIzRWRbMQA8PUKbBY1sbHxKCeX1UyEVyPXt80eyZKiuzZqpon-fecb9sfW4jGO~~JP5-5gn19cwsdpa5F9WZEVf--UufwdoVJQuqcXAPq15qHMxt4blsOwDxNc1C4fESfW2r~QI7xsVpNaINFAW~zebPgam5ZwjcMd2oIHt8oj7VoXx5qMgVUPc1iat498EhstLjxk-aeWX01Yuf2Da30mPUVhAIYy7gt5sX7jbd2xDxxXdsevKj-Se6QDxsLVd4akOJR7gcvtK3TjMUMtf0OJJFMH7JXW1Zt6bE6rnHevX70vKlREKRyPWdyd1nBgaZ~A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Cardiac glycosides suppress HIF-1α expression in vitro and in vivo. (A) Western blot showing hypoxia induction of HIF-1α in MGG123 cells and its abrogation by digoxin [D] and ouabain [O] (1 μmol/L). N, normoxia. Actin was used as a loading control. (B) IHC for HIF-1α on sections derived from an in vivo pharmacodynamic study testing systemic digoxin treatment. Upper panels show results obtained in 5 mice in the vehicle group (V1–V5); bottom panels show matching results from 5 mice in the digoxin group (D1–D5). B, brain; T, tumor. Scale bar = 100 μm. (C) Table summarizing the HIF-1α immunopositivity status in all the animals.
Evidence supporting the role of HIF-1α in cancer angiogenesis, progression, and resistance has stimulated research efforts directed at developing therapeutics targeting HIF-1α directly or indirectly ( 23, 39–41 ). Among numerous compounds shown to disable the function of HIF-1α and its pathway, cardiac glycosides such as digoxin and ouabain are unique in that they are capable of inhibiting HIF-1α protein synthesis ( 42 ). We therefore tested whether digoxin and ouabain can block hypoxia-induced HIF-1α overexpression in MGG123 cells. Both cardiac glycosides at 1 μmol/L abrogated HIF-1α protein expression by MGG123 cells under hypoxic conditions (Fig. 5A).
Treatment With Digoxin Decreased HIF-1α Expression in Orthotopic MGG123 Xenografts
The observation that our MGG123 GBM model recapitulates the hypoxic microenvironment of human GBM renders this model a suitable platform to test novel drugs that target HIF-1α. Therefore, we set out to assess whether digoxin suppresses HIF-1α and downstream effects in intracerebral MGG123 xenografts. SCID mice that each harbored a large intracerebral MGG123 xenograft were given daily intraperitoneal injections of digoxin for 5 days before death. HIF-1α IHC on brain sections showed that all 5 tumors derived from the vehicle-treated group contain areas positive for HIF-1α staining, with 4 tumors being immunopositive in central areas and 1 tumor at the periphery (Figs. 5B, C). In contrast, 4 of 5 tumors derived from digoxin-treated mice were negative for HIF-1α staining, and only 1 tumor had a HIF-1α-weakly positive area at tumor periphery (Figs. 5B, C).
Digoxin Inhibits Tumor Angiogenesis and Extends Animal Survival in the MGG123 Orthotopic Model
HIF-1α promotes angiogenesis by activating the expression of genes such as VEGF . We wanted to test whether short-term inhibition of HIF-1α expression would have an impact on tumor vasculature. CD34 IHC on the orthotopic tumors having 5-day digoxin treatment (as in Fig. 5) revealed that the densities of CD34-positive vasculature were significantly decreased in tumors derived from digoxin-treated mice compared with the vehicle-treated group (p = 0.002, Figs. 6A, B). This effect was associated with reduced levels of VEGFA mRNA in the digoxin-treated tumors compared with the vehicle-treated tumors (p = 0.01, Fig. 6C).
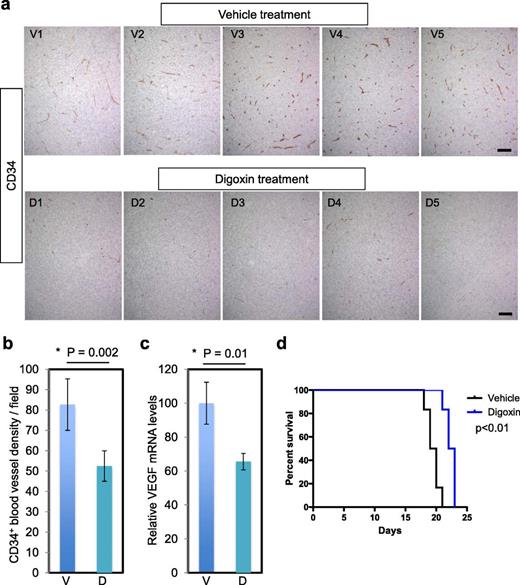
Digoxin treatment suppresses angiogenesis and extends overall survival in mice. (A) IHC for CD34 in the orthotopic tumor tissues derived from vehicle-treated mice (n = 5, V1–5) and digoxin-treated mice (n = 5, D1–5). These are the same tumors as in Figure 5. Scale bar = 100 μm. (B) Quantification of CD34-positive blood vessels showing significant reduction of tumor vessel densities in the digoxin group. (C) Quantitative RT-PCR analysis of VEGFA transcript levels in MGG123 xenografts after treatment with vehicle and digoxin. Values were normalized to the average of the vehicle group to show relative levels. V, vehicle; D, digoxin; bars, SD in (B) and (C) . (D) Kaplan-Meier analysis showing survival of mice bearing orthotopic MGG123 xenografts after treatment with vehicle or digoxin. N = 6 per group. 10 5 cells were implanted to generate tumors.
Finally, we asked whether the effects on HIF-1α and angiogenesis mediated by digoxin translate into efficacy. Mice bearing intracerebral MGG123 tumors were treated systemically with digoxin or vehicle. MGG123 grew aggressively and all mice in the vehicle group died by Day 21 post implantation. Compared with the control group, the digoxin-treated group had a modest but significant increase in overall survival (median survival time, 19.5 vs 22.5 days, p = 0.0018, Fig. 6D). We repeated this experiment and found that digoxin induced a similar significant extension in survival (median survival time, 25 vs 28 days, p = 0.01, Figure, Supplemental Digital Content 3, Supplementary Data ).
Discussion
Necrosis is one of the important diagnostic criteria distinguishing GBM (WHO grade IV) from WHO grade III anaplastic astrocytoma, and its association with poor prognosis has been well documented ( 5–7 ). The most striking pathologic characteristic of the MGG123 model is its profuse necrosis formation. Traditional GBM models based on established human cell lines rarely display necrosis ( 33, 43, 44 ). The commonly used U87 (a.k.a. U87MG) model shows a noninfiltrative growth pattern with a well-demarcated tumor mass. U87 GBM has robust neovascularization and contains no necrotic foci, revealing a key dissimilarity from human GBM ( 43–45 ). The U251 model has been shown to recapitulate some of the key features of GBM, including invasion and occasional necrosis, but does not appear to generate palisading necrosis ( 45–47 ). We and others reported a unique ability of patient-derived stem-like GBM cells to recapitulate the pathologic hallmarks of human GBM such as extensive invasion, diffuse infiltration, and hypervascularity upon orthotopic engrafting ( 34, 35, 48, 49 ). We observed necrosis ( 34 ), but assessment of 19 patient-derived GBM models in this study revealed necrosis only in 3 tumors that display semi- to noninvasive tumor phenotypes, and necrotic foci in the 2 models (MGG4 and BT74) were small and very few within the tumor. Giannini et al ( 50 ) reported robust necrosis formation in their subcutaneous xenografts of patient-derived GBM, whereas orthotopic engraftment produced necrosis much less frequently, suggesting that tumor locations or sizes are potential factors influencing necrosis generation in mice.
The transcription factor HIF-1α senses low oxygen, regulates expression of numerous genes, and promotes tumor angiogenesis, invasiveness, resistance, and stemness ( 11, 12, 51 ). In GBM, HIF-1α is not uniformly expressed within the tumor but is heterogeneously and predominantly expressed in palisading neoplastic cells that surround necrotic foci ( 9, 25, 26 ), indicating that these cells are hypoxic. Recapitulating this HIF-1α distribution pattern, the orthotopic MGG123 model demonstrated HIF-1α immunopositivity with close anatomical relationship with necrosis. Preferential gene expression by perinecrotic GBM cells was previously shown by Dong et al ( 52 ), who employed laser-capture microdissection to identify genes such as POFUT2 and PTDSR that have values as prognostic markers. Immunofluorescence detection of hypoxic cells and CD34-positive blood vessels revealed that certain distances (i.e. >60 μm) separate hypoxic areas and blood vessels. This finding indicates the heterogeneous status of oxygenation within the xenografts, mostly determined by vascular distribution, which likely contributes to necrosis formation.
HIF-1α-positive tumor cells adjacent to necrosis are as proliferative as HIF-1α-negative viable tumor cells, as illustrated by the observed high MIB-1 labeling index. Immunohistochemical analysis for stem cell markers showed that while expression of nestin and CD44 was diffusely positive throughout the tumor irrespective of relationship with blood vessels or necrosis, Sox2 positivity was predominantly observed in perinecrotic and perivascular areas. This observation is in line with the concept of the perivascular and hypoxic niche microenvironment that promotes and sustains stemness of subpopulations of GBM cells ( 18–21 ) and suggests that Sox2 might mark GBM stem cells in this model.
The ability to recapitulate GBM-relevant intratumoral hypoxia renders this model particularly suited for investigations of tumor hypoxia biology and therapeutic targeting of hypoxic cells. HIF-1α is considered an attractive target for intervention given its roles in multiple aspects of tumor malignancy such as angiogenesis and resistance ( 10, 14, 23, 40, 53 ). Compounds inhibiting HIF-1α function through different mechanisms of action are at preclinical and clinical development stages for cancer therapy, including galdanamycin and tanespimycin by enhancing proteosomal degradation of HIF-1α ( 54, 55 ); echinomycin inhibiting HIF-1α DNA binding activity ( 56, 57 ); YC-1 blocking HIF-1α expression through activating guanylate cyclase ( 58 ); and flavopiridol, which inhibits HIF-1α mRNA transcription ( 59 ). Chou et al ( 60 ) tested YC-1 as an inhibitor of HIF-1α in an orthotopic U87 GBM model in mice and showed that YC-1 prolongs survival and enhanced the efficacy of the alkylating agent BCNU.
Cardiac glycosides such as digoxin and ouabain are another class of HIF-1α inhibitor that potently suppresses the synthesis of HIF-1α ( 42 ). Zhang et al ( 42 ) demonstrated the efficacy of digoxin in blocking subcutaneous growth of B cell-lymphoma and prostate cancer xenografts, which was associated with the decrease of HIF-1α levels. Digoxin also inhibits growth of cultured GBM cells and flank GBM xenografts with concomitant reduction of HIF-1α and CD133 levels ( 18 ). Using the MGG123 model, we show that digoxin potently shuts down the HIF-1α protein expression even after its induction with hypoxic conditions in vitro. Furthermore, we demonstrate for the first time digoxin-mediated HIF-1α silencing in orthotopic GBM xenografts. This was associated with a reduction of VEGFA mRNA encoding a HIF-1α downstream angiogenic factor, and of tumor angiogenesis. These effects were translated into slight but significant extension of survival of mice bearing the aggressive MGG123 GBM. Given extensive clinical use of digoxin for the treatment of heart failure, our findings have immediate translational implications and support further investigations toward efficacy validation and a better understanding of underlying mechanisms.
In summary, the MGG123 model faithfully recapitulates the pathophysiology of the hypoxic tumor microenvironment as well as the histopathologic phenotypic hallmarks of human GBM such as invasiveness, stemness, neoangiogenesis, and proliferative activity. These features of this preclinical model allow us to study the pathophysiology of hypoxia in GBM. Furthermore, the model offers a reliable platform to test novel and existing compounds interfering with the tumor hypoxic microenvironment and angiogenesis or targeting HIF-1α to determine their therapeutic efficacy and to identify hypoxia-related biomarkers. These investigations using the clinically relevant MGG123 model might ultimately help improve the current care of patients with GBM.
References
Supporting Information
Author notes
Sources of support: This work was supported by a grant from the National Institute of Health (Grant Number R01NS032677) and the ABTA discovery grant.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Web site ( www.jneuropath.com ).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}