





Abstract
Lysosomal storage disorders constitute a large group of genetic diseases, many of which are characterized by mental retardation and other neurologic symptoms. The mechanisms of neural dysfunction remain poorly understood. Because neural progenitor cells (NPCs) are fundamentally important to normal brain development and function, we investigated NPC properties in a canine model of mucopolysaccharidosis VII (MPS VII). MPS VII is a lysosomal storage disorder characterized by defects in the catabolism of glycosaminoglycans. NPCs were isolated from the olfactory bulb, cerebellum, and striatal subventricular zone of normal and MPS VII (β-glucuronidase-deficient) postnatal dog brains. Canine NPCs (cNPCs) from normal and MPS VII brains had similar growth curves, but cerebellar-derived cNPCs grew significantly slower than those derived from other regions. In differentiation assays, MPS VII cNPCs from the striatal subventricular zone and cerebellum generated fewer mature neuronal and/or glial cells than normal, and MPS VII olfactory bulb-derived cNPCs retained significantly more phenotypically immature cells. These differences were only present at the earliest time point after isolation; at later passages, there were no differences attributable to genotype. The data suggest that MPS VII cNPCs respond differently to developmental cues in vivo, probably because of the diseased neural microenvironment rather than intrinsic cellular deficits.
Introduction
One approach to understanding the effects of neurogenetic disease is to examine properties of neural progenitor cells (NPCs). Neural stem cells are multipotent and capable of extended self-renewal, whereas NPCs exhibit more restricted potency (1,2). The ability to propagate NPCs in vitro affords the opportunity to investigate neuropathogenic mechanisms of neurodegenerative diseases, but there are few studies investigating the role of NPCs in neurologic diseases (3-7).
The lysosomal storage disorders (LSDs) are monogenic diseases that feature defects in the degradation of macromolecules, leading to accumulation of excess substrates in cells. Most of the LSDs involving the CNS manifest as mental retardation. Despite a clear understanding of the normal functions of lysosomal enzymes and the histopathologic sequelae, the cellular and molecular mechanisms underlying neural dysfunction are poorly understood (8-10).
We investigated NPCs in a canine model of mucopolysaccharidosis (MPS) type VII (Sly disease), a deficiency of β-glucuronidase, which results in lysosomal accumulation of glycosaminoglycans (11-13). Glycosaminoglycans and glycosaminoglycan-containing proteoglycans are involved in the maintenance of the stem cell niche, as well as in modulation of neuronal migration and axonal guidance (14-20). There are no gross defects in neural structures attributed to the accumulation of substrate during development; thus it is likely that accumulating substrates affect optimal functioning of the postnatal brain.
NPCs from unaffected and MPS VII dogs were examined in vitro for intrinsic functional defects attributable to genotype. Multipotent NPCs reside in the subventricular zone (SVZ), olfactory bulb, and hippocampus (21-27), as well as within the white matter tracts of the cerebellum (28-31). However, there have been very few studies in which the properties of NPCs from different brain regions were directly compared (32,33). We chose to evaluate canine NPCs (cNPCs) derived from the olfactory bulb and cerebellum because both sites are surgically accessible in humans and dogs for potential autologous transplantation therapy. We compared them with canine SVZ cells, because the SVZ has been well characterized for neural stem cells in rodents and humans (24,25,27,34-38). Significant differences were found between normal and diseased cNPCs from all 3 regions. However, differences were significant only at the earliest time point after explantation, implicating the diseased microenvironment of the brain rather than intrinsic cellular changes.
Materials and Methods
Experimental Animals
Dogs were raised in the animal colony at the University of Pennsylvania School of Veterinary Medicine according to National Institutes of Health and U.S. Department of Agriculture guidelines for the use of animals in research, and the study was approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. Screening for MPS VII dogs was performed by evaluating Wright-Giemsa-stained blood films for the presence of storage granules in white blood cells. Normal, carrier, or MPS VII genotype was then established by polymerase chain reaction for the mutation (39).
Isolation of Canine Neural Progenitor Cells
Three unaffected and 4 MPS VII-affected dogs ranging in age from 19 to 23 days were humanely killed by intravenous injection of a barbiturate solution. The brain was removed, placed into a balanced salt solution, and then dissected grossly. The olfactory bulbs, cerebellum (lobule and vermis), and rostral one third of the brain (located just caudal to the olfactory bulbs and rostral to the hippocampus) were separated from the brain and from each other. The rostral one third of the brain was further dissected to leave primarily the tissue surrounding the lateral ventricles (including parts of the corpus callosum, caudate putamen, and septae). The resultant tissues were minced separately and then digested in 0.25% trypsin (Worthington, Lakewood, NJ). The enzymatic digestion was stopped with addition of fetal bovine serum (FBS) (Hyclone, Logan, UT), and tissues were then incubated with DNase I (Sigma, St. Louis, MO) before trituration to a single cell suspension. Cell suspensions were centrifuged at 700 rpm at 4°C, resuspended in 10% FBS plating medium (see below), and triturated. The total number of viable cells was determined by manual count on a hemacytometer; cell viability was assessed using trypan blue exclusion (0.4%; Sigma).
Canine Neural Progenitor Cell Culture and Expansion
cNPCs were plated into 25-cm2 tissue culture flasks (Corning, Acton, MA) coated with 10 μg/mL poly-D-lysine (Sigma) at a concentration of 4 × 104/cm2 in plating medium for 24 to 48 hours. Plating medium consisted of Dulbecco's modified Eagle's medium/F-12 (1:1 ratio; Invitrogen, Carlsbad, CA) supplemented with 10% FBS, 1% N2 supplement (Invitrogen), 1% antibiotic-antimycotic (100 U/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B; Invitrogen), and 1% L-glutamine (2 mM; Invitrogen). After 24 to 48 hours, the medium was changed to a serum-free feeding medium consisting of Dulbecco's modified Eagle's medium/F-12 supplemented with 1% N2 supplement, 1% antibiotic-antimycotic, and 1% L-glutamine. The standard combination of growth factors consisted of 20 ng/mL epidermal growth factor (recombinant murine; Roche, Nutley, NJ), 20 ng/mL basic fibroblast growth factor (FGF) or FGF-2 (recombinant human; Promega, Madison, WI), and heparin (5 μg/mL; Sigma). cNPC cultures were maintained at 37°C in humidified 5% CO2 tissue culture incubators and fed every 3 to 5 days by changing half of the medium and adding fresh growth factors. Cultures were passaged at ~90% confluence by trypsinizing (0.05% trypsin-EDTA; Invitrogen) and replating at a concentration of 4 × 104/cm2.
Immunocytochemistry
Cultures were differentiated by plating in the absence of growth factors in an 8-well chamber slide (Lab-Tek II; Nalge Nunc, Rochester, NY) at a concentration of 4 × 104 cells/well in feeding medium supplemented with 1% FBS. Undifferentiated cNPCs were plated at a concentration of 2 × 104/well onto poly-D-lysine-coated 8-well glass slides (Cel-Line, Erie Scientific, Portsmouth, NH) and allowed to attach overnight in feeding medium containing growth factors. Differentiated and undifferentiated cNPC cultures were processed for immunocytochemistry after 10 to 12 days and 24 hours, respectively. In some cases, retinoic acid (1 μM; Sigma), platelet-derived growth factor (10 ng/mL; Sigma), or 10% FBS was added to feeding medium in the absence of growth factors to promote differentiation.
The polyclonal primary antibodies used were rabbit polyclonal anti-nestin, 1:60 dilution (rabbit 130; kind gift of R. McKay, National Institutes of Health) and rabbit polyclonal anti-glial fibrillary acidic protein (GFAP), 1:100 dilution (Chemicon, Temecula, CA). Primary monoclonal antibodies consisted of rat anti-GFAP, 1:1 dilution (IgG; kind gift of V. Lee, University of Pennsylvania); mouse anti-β-tubulin III, 1:300 dilution (IgG; Chemicon); mouse anti-MAP2ab, 1:300 dilution (IgG; Chemicon); mouse anti-O4, 1:3 dilution (IgM); and mouse anti-galactocerebroside, 1:1 dilution (IgG3) (both oligodendrocyte markers were generous gifts of J. Grinspan, Children's Hospital of Philadelphia). Secondary fluorescent antibodies used were goat anti-mouse IgG/IgM fluorescein isothiocyanate, 1:300 dilution (Chemicon); goat anti-rabbit IgG 594 Alexa Fluor, 1:300 dilution (Molecular Probes, Eugene, OR); goat anti-rabbit IgG 488 Alexa Fluor, 1:300 dilution (Molecular Probes); and goat anti-mouse IgM Alexa Fluor 488, 1:300 dilution (Molecular Probes).
For all intracellular markers, cells were rinsed in Tris-buffered saline (50 mM Tris-base and 0.15 M NaCl, pH 7.6), fixed in 4% paraformaldehyde (Sigma), blocked in 5% goat serum (Invitrogen) with 0.1% Triton X-100 (Sigma), and then incubated with primary antibody in 1% goat serum with 0.02% Triton X-100. The secondary antibody was applied for 1 hour at room temperature or overnight at 4°C. Cell surface marker staining was performed on live cells. All slides were mounted in Vectashield containing 4′,6 diamidino-2-phenylindole (Vector Laboratories, Burlingame, VT).
Quantitation of Immunofluorescence
A minimum of 10 fields for each marker were photographed using a SPOT RT camera (Diagnostic Instruments, Sterling Heights, MI). An effort was made to sample representatively; when differentiation was not uniformly distributed, similar numbers of fields without positive staining were analyzed. The 4′,6 diamidino-2-phenylindole-stained nucleus was used to count the total number of cells in all fields using a manual tag with Image-Pro Plus software (version 4.0; Media Cybernetics, Silver Spring, MD). The percentage of cells that stained positively for an immunofluorescent marker was obtained by averaging the percentage of positive cells for all fields.
Canine Neural Progenitor Cell Population Parameters and Statistics
For each cell culture, the numbers of cells plated at the beginning and harvested at the end of each passage were recorded. Cultures were plated in triplicate for comparison of unaffected and MPS VII NPCs from the cerebellum, olfactory bulb, and SVZ. The total potential cell number generated was calculated by multiplying the total number of viable cells harvested after each passage by the ratio of the total cells harvested to the total cells plated. The product for each passage was added to the subsequent passage to produce the total potential cells generated if all cells harvested were plated (40). Cell doubling time was determined from the standard formula: doubling time = [(log P − log H)/log 2]/D; where P is the number of cells plated, H is the number of cells harvested, and D is the days between plating and harvest (5).
Statistical analysis was performed to determine whether there was a difference in population doubling times and differentiation ability between cNPCs derived from MPS VII and unaffected dogs in each region using an unpaired Welch t-test or a Mann-Whitney test. Comparisons between the growth parameters of multiple brain regions were performed using the Kruskal-Wallis nonparametric analysis of variance and, where appropriate, Dunn's multiple comparisons test.
Results
Comparison of Canine Neural Progenitor Cell Growth Parameters From 3 Different Brain Regions of Mucopolysaccharidosis VII-Affected and Unaffected Dogs
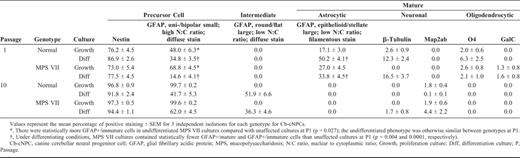
The cNPCs were selected by epigenetic stimulation in a serum-free medium and grown as an adherent monolayer on a poly-D-lysine substrate in the presence of epidermal growth factor and FGF-2 with heparin. There were no apparent differences in the morphology of cNPCs in culture regardless of disease phenotype or region of derivation (Fig. 1). The cells were primarily bipolar or unipolar and fusiform and grew in a lacy, interconnected fashion. Neurospheres occasionally formed from dense adherent cell clusters and floated free or remained attached to the cellular substratum. The populations became more morphologically homogeneous with successive passages.
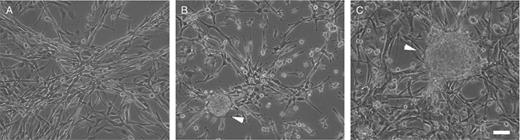
Canine neural progenitor cell (cNPC) morphology in culture. Phase-contrast photomicrographs at the primary passage for mucopolysaccharidosis VII cerebellar cNPCs (A), unaffected olfactory bulb cNPCs (B), and unaffected subventricular zone cNPCs (C). Arrowheads point to neurospheres, which were present in varying numbers in cultures from all sites. Scale bar = 25 μm.
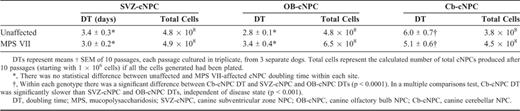
Cultures were established in triplicate for each region from MPS VII and normal dogs (n = 3). Because NPC proliferation in response to FGF-2 is potentiated by cell surface heparan sulfates (41-43), defects in heparan sulfate catabolism may alter growth parameters. We therefore evaluated the population doubling times over the course of 10 passages for each region of origin (Table 1). We also determined the total potential numbers of cells generated if all cells had been plated after each passage Table 1).
Comparison of Unaffected and MPS VII-Affected Growth Parameters in cNPCs Derived From 3 Different Brain Regions
Comparison of Unaffected and MPS VII-Affected Growth Parameters in cNPCs Derived From 3 Different Brain Regions
Most of the cNPCs were actively proliferating at the end of passage (P) 10, corresponding to ~80 days in vitro. Population doubling time for actively proliferating cultures ranged from 1 to 18 days. There was no statistical difference in population doubling or total cell expansion number between MPS VII and unaffected cNPC cultures (p = 0.1-0.3). The doubling time for cerebellar-derived cNPC cultures was significantly slower compared with those from the SVZ and olfactory bulb. This observation was true for both unaffected and MPS VII-affected cNPCs.
Immunophenotype and Morphology of Undifferentiated Canine Neural Progenitor Cell Cultures
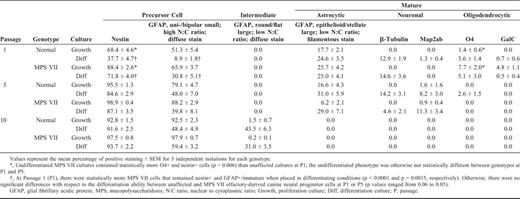
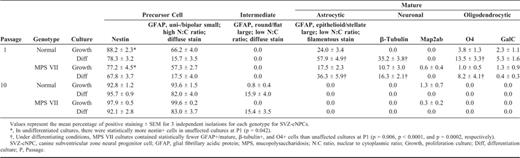
Aliquots of cultures from each genotype and from each region were plated overnight in the presence of growth factors for analysis of the undifferentiated phenotype. Immunophenotyping was performed for all regions at P1 and P10; olfactory bulb-derived cNPCs (OB-cNPCs) were also assessed at P5 (Tables 2–4).
Undifferentiated and Differentiated Immunophenotypes of Canine Olfactory Bulb Neural Progenitor Cells
Undifferentiated and Differentiated Immunophenotypes of Canine Olfactory Bulb Neural Progenitor Cells
GFAP is a marker of both differentiated astrocytes and neural stem cells (36, 37, 44, 45), whereas nestin is used as a marker of neural progenitor cells (46-48). The neural stem cells of the SVZ are reported to be both nestin+ and GFAP+ (35). The undifferentiated cNPC phenotype was similar for both unaffected and MPS VII cNPC cultures and for all regions; the majority of cells stained positively for GFAP and nestin. The GFAP+ cells were divided into 2 groups based on cellular morphology and staining. Cells with the following characteristics were considered "immature" or undifferentiated: small nucleus; unipolar, bipolar, or round shape with a high nuclear to cytoplasmic ratio (N:C ratio); and a nonfilamentous, diffuse GFAP staining pattern. In contrast, cells with large nuclei, a stellate or flattened, fibroblast-like morphology with a low N:C ratio, and filamentous staining pattern were considered "mature" because these morphologies are consistent with mature type 1 and type 2 astrocytes (49) (Fig. 2). At the latest time evaluated (P10), a third, intermediate type of GFAP+ cell was observed, but only in cultures that had been subjected to differentiating conditions. This type had a mature fibroblast-like morphology-epithelioid and flattened with a low N:C ratio-but had a diffuse GFAP staining pattern, rather than a mature filamentous one (Fig. 2).
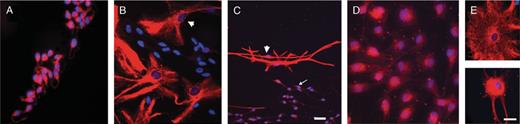
Glial fibrillary acidic protein (GFAP)+ cells showed 3 types of morphology and staining. (A) The majority of cells grown in the presence of growth factor were small, round, or fusiform, and stained diffusely for GFAP. (B) Some cells grown under differentiating conditions developed the morphology and filamentous GFAP staining of mature type 1 astrocytes (arrowhead). (C) Both immature (arrow) and mature (arrowhead) GFAP+ cells were present in differentiation cultures. (D) At P10, GFAP+/mature cells had a diffuse staining pattern. (E) Comparison of filamentous (top panel; P5) and diffuse (bottom panel; P10) staining in GFAP+ cells with mature type 1 astrocyte morphology. Scale bar = (A, B, D, E) 25 μm; (C) 50 μm.
Spurious GFAP staining with polyclonal GFAP antiserum has been reported, probably due to vimentin or nestin cross-reactivity (50). To verify that GFAP staining in cNPC cultures was specific, we costained both undifferentiated and differentiated cultures with a polyclonal rabbit GFAP antiserum and a rat monoclonal anti-GFAP antibody. The polyclonal and monoclonal anti-GFAP antibodies stained the same cells (data not shown). In addition, the same 2 types of staining patterns in immature and mature cells-diffuse and filamentous, respectively-were observed with both polyclonal and monoclonal antibodies.
In the undifferentiated cultures, as passage number increased, the proportion of mature GFAP+ cells decreased and immature GFAP+ cells and nestin+ cells increased (Tables 2–4). For the neuronal markers (β-tubulin and Map2ab) and the oligodendrocyte markers (O4 and galactocerebroside), a small proportion of positive staining was seen at P1. Over successive passages, the cultures maintained in growth factors developed a more homogeneous, immature phenotype with the exception of Map2ab staining. At P5 and P10, there was some Map2ab-staining (<2% of cells) in the undifferentiated cultures from the olfactory bulb and the SVZ and cerebellum, respectively. The proportion of Map2ab+ cells in undifferentiated cultures reported in Tables 2–4 represents Map2ab+ cells with appropriate neuronal morphology. In addition to Map2ab+ mature cells, there were Map2ab+ cells with an immature morphology (small unipolar or bipolar cells with a high N:C ratio) (Fig. 3). The proportion of Map2ab+ cells with immature morphology in proliferating (undifferentiated) cultures increased with passage number for cerebellar-derived cNPCs, but remained consistent for SVZ-derived cNPCs (Table 5). OB-cNPC cultures contained Map2ab+ immature cells only at P5.
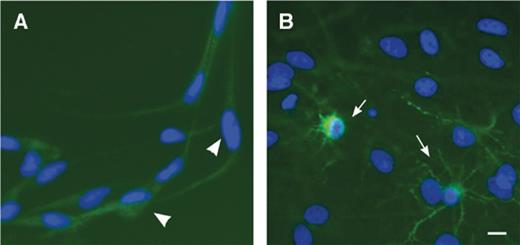
Morphology of Map2ab+ cells in canine neural progenitor cell cultures. (A) In the presence of growth factors, small Map2ab+ cells are noted with bipolar or unipolar morphology (arrowheads). (B) Upon differentiation by withdrawal of growth factors and addition of serum, variably sized cells with multiple processes are observed (arrows). Scale bar = 20 μm.
Immunophenotype and Morphology of Differentiated Canine Neural Progenitor Cell Cultures
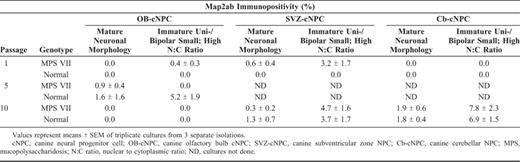
For analysis of the differentiated phenotype, aliquots were plated for 10 to 12 days in 1% serum-supplemented medium in the absence of growth factors. Removal of growth factors resulted in proportions of cells that stained positively for neuronal, astrocytic, and oligodendrocytic markers and displayed a phenotype-appropriate morphology (Tables 2-4; Fig. 4). The greatest amount of differentiation was observed at the earliest passages evaluated. At P1 and P5, markers of differentiated neurons, astrocytes, and oligodendrocytes were observed for all cultures evaluated. In addition, the proportions of nestin+ and immature GFAP+ cells tended to be lower than that of the undifferentiated (growth factor-maintained) phenotype for the same passages.
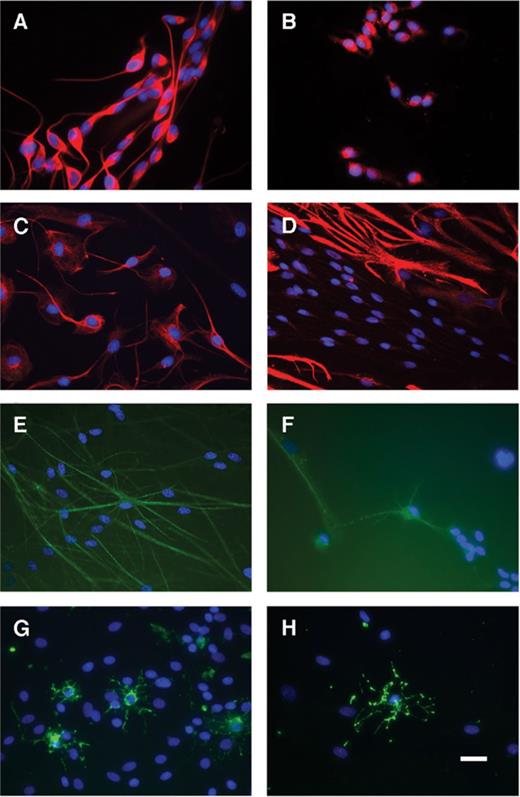
Immunophenotyping of olfactory bulb-derived canine neural progenitor cells (OB-cNPCs). (A, B) Undifferentiated cultures. The majority of cells stained positively for nestin (A) and glial fibrillary acidic protein (GFAP) (B). Cells are uniformly unipolar or bipolar with a simple, immature morphology. (C--H) Differentiated cultures (growth factor withdrawal). The intensity of nestin staining (C) decreases and mature cellular morphology is noted for both nestin+ (C) and GFAP+ (D) cells. (E--H) Some OB-cNPCs stain positively for neuronal markers β-tubulin III (E) and Map2ab (F) and oligodendrocytic markers O4 (G) and GalC (H)). The appropriate morphology for astrocytes (D), neurons (E, F), and oligodendrocytes (G, H) is observed. Scale bar = 25 μm.
At P10, however, there was a decrease in differentiation after growth factor withdrawal that was uniform across cNPCs regardless of disease status and site of origin. Few to no cells stained positively for neuronal and oligodendrocytic differentiation markers. Relative to earlier passages, the proportion of immature GFAP+ cells and nestin+ cells increased. Moreover, there was a disparity between cellular morphology and staining pattern. Even GFAP+ with mature morphology consistently stained with an immature, diffuse pattern rather than a filamentous one. To determine whether the lack of differentiation at P10 was a function of the passive method of differentiation (withdrawal of growth factors), we attempted to potentiate differentiation with retinoic acid or platelet-derived growth factor (51,52), but neither potentiated differentiation in P10 OB-cNPC cultures. Similarly, there was no effect on P1 OB-cNPC cultures (data not shown). Differentiation of OB-cNPC cultures in 10% FBS without growth factors increased the proportion of β-tubulin III+ cells in P1 cultures relative to passive differentiation cultures but had no effect on P10 OB-cNPC cultures (data not shown).
Unaffected and MPS VII cNPCs were evaluated for differences in phenotypes upon differentiation. The only statistical differences in differentiation phenotype between genotypes were present at the earliest passage evaluated after isolation, P1. The overall tendency of difference was for MPS VII cNPCs to favor an immature phenotype. At P1, more MPS VII OB-cNPCs remained nestin+ and GFAP+/immature when placed in differentiating conditions (p < 0.0001 and p = 0.0015, respectively), the MPS VII SVZ-cNPC cultures contained fewer GFAP+/mature, β-tubulin+, and O4+ cells than unaffected cultures (p = 0.006, p < 0.0001, and p = 0.0002, respectively), and in MPS VII cerebellum-derived cNPC differentiation cultures, there were fewer GFAP+/mature and GFAP+/immature cells than in unaffected cultures (p = 0.004 and 0.0001, respectively).
Discussion
In MPS VII, β-glucuronidase deficiency results primarily in lysosomal accumulation and excretion of excessive heparan, dermatan, and chondroitin sulfates (11-13). Both heparan and chondroitin sulfate proteoglycans are implicated in the response of NPCs to morphogens, maintenance of the stem cell niche, and modulation of neuronal migration and axonal guidance (14-20). Although there are no gross defects in neural structures attributed to the accumulation of substrate during development, it is possible that there are postnatal effects of the accumulating substrates. The data shown here indicate that abnormal substrate accumulation results in aberrant NPC function. In our earlier studies using the mouse model of MPS VII, we found no evidence that disease phenotype affected either in vitro growth rate or neuronal differentiation of neonatal day 3 murine NPCs (5). Developmentally, a postnatal day 3 mouse brain is comparable to a human fetal brain in the early second trimester, whereas a 21-day-old dog is approximately equivalent to a human fetus in its early third trimester (53). Thus, the diseased cNPCs in this study represent a later developmental stage. Similar to the mouse model, our data did not show any differences between diseased and unaffected cNPCs with respect to in vitro growth potential. However, when subjected to differentiation conditions, phenotypic differences were present between MPS VII and normal cNPCs. Differences associated with disease genotype were seen only in cultures at the earliest point after isolation. The MPS VII mouse NPC studies were performed at P12 to 14 (5), so it is possible they may show similar differences immediately after isolation. The increasing similarity in immunophenotype between diseased and normal cNPC cultures over time suggests that MPS VII cNPCs can respond as normal cNPCs to growth factors in vitro.
Appropriate models for transplantation therapy before clinical human trials are essential. Whereas rodents are invaluable models for many neurologic diseases, the differences in size and architecture of the murine and human brain are vast. The MPS VII dog model is therefore a better paradigm for therapeutic transplantation strategies than the mouse. In this study we determined that postnatal cNPCs could be expanded ex vivo for at least 10 passages (~80 days), producing up to a 600-fold increase in cell number. This level of expansion would permit generation of sufficient cells for transplantation into a brain the size of an adult dog (or a 1-year-old child). The model would be to obtain a biopsy from the cerebellum or olfactory bulb, transduce the cells early in culture with an integrating gene transfer vector (e.g. lentivirus) expressing the normal β-glucuronidase cDNA, expand the corrected cells in vitro, and transplant the autologous, genetically corrected cells into the patient's brain. The potential advantage of using NPCs is that they can migrate within the brain to provide secreted lysosomal enzyme to the globally distributed lesions. cNPC expansion is similar to that reported for human embryonic NPCs, which were expanded 102-fold after approximately 50 to 70 days; a 107-fold expansion required nearly 1 year of propagation ex vivo (54). In contrast, mouse embryonic NPCs propagated over a similar time period for the same number of passages produced a 107-fold increase in cells (40).
The doubling time for diseased and normal postnatal cNPCs was similar to that obtained for postnatal mouse and human embryonic NPCs. Mean postnatal cNPC doubling times ranged from 2.8 to 6.0 days. Mean doubling times for postnatal mouse NPCs ranged from 3.2 to 9.0 days and for human embryonic NPCs ranged from 7 to 10 days (5,54). Although cNPC mean doubling times were comparable to those of human and rodent NPCs, there was a difference between growth rates from different sites. Our findings support the use of olfactory bulb cNPCs over cerebellar-derived cNPCs for autologous transplantation therapy. Olfactory bulb cNPCs had a shorter expansion time and generated greater numbers of cells.
The undifferentiated phenotype of diseased and normal cNPCs was largely nestin+ and GFAP+. A previous study reported that undifferentiated cNPCs grown with epidermal growth factor were nestin+, but did not evaluate GFAP expression (55). Although GFAP+ cells have been shown to be stem cells in vivo (36,37,56-58), reports of GFAP staining in undifferentiated human and mouse NPCs ranges from absent to high, depending upon the method of cell culture (5,22,54,59-62), whereas nestin immunoreactivity is consistently high in all reports. In all undifferentiated cNPC cultures, the presence of small numbers of Map2ab+ cells with an immature morphology was an unexpected finding. While there were Map2ab+ cells with neuronal morphology that represented differentiated cells, the majority of Map2ab+ cells had an immature morphology. It has been shown previously that subsequent to CNS injury, reactive astrocytes express neuronal antigens Tau, Map2, neuron-specific enolase, and GABA in addition to GFAP and nestin (63-66). Reactive astrocytes may represent astrocytes that revert to bipotential progenitors (cells that show both neuronal and glial phenotypes) or may be recruited from neural stem cells (65,67). In vitro, primary astrocytes derived from mouse brain are nestin+ and GFAP+, but show no immunoreactivity to neuronal antigens Map2 and Tau; however, astrocytes derived from NPCs are GFAP+, nestin+, Map2+, and Tau+ (65). In support of these findings, cortical injury studies with nestin promoter/lacZ transgenic mice show that nestin+ cells migrate from the subventricular zone to the site of injury (63,67). Thus, the Map2ab+ cells with immature morphology in undifferentiated cNPC cultures may represent a cell type analogous to reactive astrocytes.
In differentiation assays for both diseased and normal cNPCs, there was markedly less immunoreactivity for mature markers and an increase in immature morphology by P10. It is possible that after prolonged growth factor exposure, withdrawal of growth factors was an insufficient stimulus for differentiation. Attempts to potentiate differentiation with retinoic acid and platelet-derived growth factor were unsuccessful for both early and late passage cNPC cultures. From a gene therapeutic standpoint, however, cNPC differentiation is not essential as long as enzyme delivery is accomplished.
These studies indicate that canine MPS VII NPCs derived from surgically accessible sites can be adequately expanded to serve as potential autologous sources of NPCs for ex vivo gene therapy and therapeutic transplantation in a brain that is scaled to that of a human child. The phenotypic differences between MPS VII and normal cNPCs shortly after isolation and their disappearance with time in culture implicate the diseased neural environment in the pathogenesis of MPS VII neurologic disease. Thus, future studies on transplantation will need to take into account whether the microenvironment into which the corrected cells are transplanted will affect the function of the genetically corrected donor cells.
Acknowledgments
We thank A. Polesky, E. Cabacungan, and T. Clarke for expert technical assistance; P. O'Donnell and Dr. M. Haskins of the Animal Models Core of the W. F. Goodman Center for Comparative Medical Genetics (RR-02512) School of Veterinary Medicine for assistance with the animals; and Drs. J. Grinspan, G. Tennekoon, M. Selzer, and G. Heuer for helpful discussions.
References
Author notes
This work was supported by National Institutes of Health Grants DK42707, DK46637, and DK63973. RMW was supported by a fellowship from the National Center for Research Resources (RR07063).

{kind=link}
{kind=link}
{kind=link}
{kind=link}